
| Amyotrophic lateral sclerosis | |
|---|---|
| Other names |
|
 | |
| The parts of the nervous system affected by ALS, highlighting the upper motor neurons, lower motor neurons, and frontotemporal regions of the brain. These cause progressive symptoms in skeletal muscles throughout the body. | |
| Specialty | Neurology |
| Symptoms |
Early: Stiff muscles, muscle twitches, gradual increasing weakness, typically in a limb Later: Difficulty in speaking, swallowing, and breathing; respiratory failure About 25-30% of cases start in the mouth + throat region. 10-15% of cases also have frontotemporal dementia |
| Complications | Falling (accident); Respiratory failure; Pneumonia; Malnutrition |
| Usual onset | 45 – 75 years |
| Causes | Unknown (most), genetic (about 15%) |
| Risk factors | Genetic risk factors including UNC13A, Ataxin-2; age; male sex; smoking; physical exercise; head injury; occupational and environmental exposures |
| Diagnostic method | Clinical diagnosis of exclusion, based on the presence of symptoms in the upper and lower motor neurons with progressive muscular weakness in which no other explanation can be found. Supportive evidence from electromyography, genetic testing, and neuroimaging |
| Differential diagnosis | Multifocal motor neuropathy, Kennedy's disease, Hereditary spastic paraplegia, Nerve compression syndrome, Diabetic neuropathy, Post-polio syndrome, Myasthenia gravis, Multiple sclerosis |
| Treatment | Walker (mobility); Wheelchair; Non-invasive ventilation;Feeding tube; Augmentative and alternative communication; symptomatic management |
| Medication | Riluzole, Edaravone; Sodium phenylbutyrate/ursodoxicoltaurine; Dextromethorphan/quinidine |
| Prognosis | Life expectancy highly variable but most often 2–4 years from diagnosis |
| Frequency | Incidence: 1.6/100,000 individuals per year; Prevalence: 4.4/100,000 living individuals Lifetime risk, 1 in 400 individuals |
Amyotrophic lateral sclerosis (ALS), also known as motor neurone disease (MND) or Lou Gehrig's disease, is a neurodegenerative disease that results in the progressive loss of motor neurons that control voluntary muscles. ALS is the most common form of the motor neuron diseases. Early symptoms of ALS include stiff muscles, muscle twitches, and gradual increasing weakness and muscle wasting.Limb-onset ALS begins with weakness in the arms or legs, while bulbar-onset ALS begins with difficulty speaking or swallowing. Around half of people with ALS develop at least mild difficulties with thinking and behavior, and about 15% develop frontotemporal dementia. Motor neuron loss continues until the ability to eat, speak, move, and finally the ability to breathe is lost.
Most cases of ALS (about 90% to 95%) have no known cause, and are known as sporadic ALS. However, both genetic and environmental factors are believed to be involved. The remaining 5% to 10% of cases have a genetic cause, often linked to a history of the disease in the family, and these are known as genetic ALS. About half of these genetic cases are due to one of two specific genes. The diagnosis is based on a person's signs and symptoms, with testing done to rule out other potential causes.
There is no known cure for ALS. The goal of treatment is to slow the disease and improve symptoms. Treatments that slow ALS include riluzole (extends life by two to three months) and sodium phenylbutyrate/ursodoxicoltaurine (extends life by around seven months). Non-invasive ventilation may result in both improved quality and length of life.Mechanical ventilation can prolong survival but does not stop disease progression. A feeding tube may help maintain weight and nutrition. Death is usually caused by respiratory failure. The disease can affect people of any age, but usually starts around the age of 60. The average survival from onset to death is two to four years, though this can vary, and about 10% survive longer than ten years.
Descriptions of the disease date back to at least 1824 by Charles Bell. In 1869, the connection between the symptoms and the underlying neurological problems was first described by French neurologist Jean-Martin Charcot, who in 1874 began using the term amyotrophic lateral sclerosis.
Classification
ALS is a motor neuron disease, which is a group of neurological disorders that selectively affect motor neurons, the cells that control voluntary muscles of the body. Other motor neuron diseases include primary lateral sclerosis (PLS), progressive muscular atrophy (PMA), progressive bulbar palsy, pseudobulbar palsy, and monomelic amyotrophy (MMA).
ALS itself can be classified in a few different ways: by which part of the motor neurons are affected; by the parts of the body first affected; whether it is genetic; and the age at which it started.
Subtypes of motor neuron degeneration

ALS can be classified by the types of motor neurons that are affected. To successfully control any voluntary muscle in the body, a signal must be sent from the motor cortex in the brain down the upper motor neuron as it travels down the spinal cord. There, it connects via a synapse to the lower motor neuron which connects to the muscle itself. Damage to either the upper or lower motor neuron, as it makes its way from the brain to muscle, causes different types of symptoms. Damage to the upper motor neuron typically causes spasticity, stiffness, brisk reflexes, and/or clonus, while damage to the lower motor neuron typically causes weakness, atrophy, and fasciculation.
"Typical ALS" or "classical ALS" involves degeneration to both the upper motor neurons in the brain and the lower motor neurons in the spinal cord.Primary lateral sclerosis (PLS) involves degeneration of only the upper motor neurons, and progressive muscular atrophy (PMA) involves only the lower motor neurons. There is debate over whether PLS and PMA are separate diseases or simply variants of ALS.
| Main ALS Subtypes | Upper motor neuron degeneration | Lower motor neuron degeneration |
|---|---|---|
| Classical ALS | Yes | Yes |
| Primary lateral sclerosis (PLS) | Yes | No |
| Progressive muscular atrophy (PMA) | No | Yes |
Classical ALS accounts for about 70% of all cases of ALS and can be subdivided into limb-onset ALS (also known as spinal-onset) and bulbar-onset ALS. Limb-onset ALS begins with weakness in the hands, arms, feet, and/or legs and accounts for about two-thirds of all classical ALS cases. Bulbar-onset ALS begins with weakness in the muscles of speech, chewing, and swallowing and accounts for about one-third of classical ALS cases. A rarer type of classical ALS affecting around 3% of patients is respiratory-onset, in which the initial symptoms are difficulty breathing (dyspnea) upon exertion, at rest, or while lying flat (orthopnea).
Primary lateral sclerosis (PLS) is a subtype of the overall ALS category which accounts for about 5% of all cases and affects only affects the upper motor neurons in the arms, legs, and bulbar region. However, more than 75% of people with apparent PLS go on to later develop lower motor neuron signs within four years of symptom onset, meaning that a definitive diagnosis of PLS cannot be made until several years have passed. PLS has a better prognosis than classical ALS, as it progresses slower, results in less functional decline, does not affect the ability to breathe, and causes less severe weight loss than classical ALS.
Progressive muscular atrophy (PMA) is another subtype that accounts for about 5% of the overall ALS category and affects lower motor neurons in the arms, legs, and bulbar region. While PMA is associated with longer survival on average than classical ALS, it is still progressive over time, eventually leading to respiratory failure and death. As with PLS developing into classical ALS, PMA can also develop into classical ALS over time if the lower motor neuron involvement progresses to include upper motor neurons, in which case the diagnosis might be changed to classic ALS.
Rare isolated variants of ALS
Isolated variants of ALS have symptoms that are limited to a single region for at least a year; they progress more slowly than classical ALS and are associated with longer survival. These regional variants of ALS can only be considered as a diagnosis should the initial symptoms fail to spread to other spinal cord regions for an extended period of time (at least 12 months). Flail arm syndrome is characterized by lower motor neuron damage affecting the arm muscles, typically starting with the upper arms symmetrically and progressing downwards to the hands. Flail leg syndrome is characterized by lower motor neuron damage leading to asymmetrical weakness and wasting in the legs starting around the feet. Isolated bulbar ALS is characterized by upper or lower motor neuron damage in the bulbar region (in the absence of limb symptoms for at least 20 months), leading to gradual onset of difficulty with speech (dysarthria) and swallowing (dysphagia).

Age of onset
ALS can also be classified based on the age of onset. While the peak age of onset is 58 to 63 for sporadic ALS and 47 to 52 for familial ALS, about 10% of all cases of ALS begin before age 45 ("young-onset" ALS), and about 1% of all cases begin before age 25 (juvenile ALS). People who develop young-onset ALS are more likely to be male, less likely to have bulbar onset of symptoms, and more likely to have a slower progression of the disease. Juvenile ALS is more likely to be familial than adult-onset ALS; genes known to be associated with juvenile ALS include ALS2, SETX, SPG11, FUS, and SIGMAR1. Although most people with juvenile ALS live longer than those with adult-onset ALS, some of them have specific mutations in FUS and SOD1 that are associated with a poor prognosis. Late onset (after age 65) is associated with a more rapid functional decline and shorter survival.
Signs and symptoms
The disorder causes muscle weakness, atrophy, and muscle spasms throughout the body due to the degeneration of the upper motor and lower motor neurons. Sensory nerves and the autonomic nervous system are generally unaffected, meaning the majority of people with ALS maintain hearing, sight, touch, smell, and taste.
Initial symptoms
The start of ALS may be so subtle that the symptoms are overlooked. The earliest symptoms of ALS are muscle weakness or muscle atrophy, typically on one side of the body. Other presenting symptoms include trouble swallowing or breathing, cramping, or stiffness of affected muscles; muscle weakness affecting an arm or a leg; or slurred and nasal speech. The parts of the body affected by early symptoms of ALS depend on which motor neurons in the body are damaged first.
In limb-onset ALS, the first symptoms are in the arms or the legs. If the legs are affected first, people may experience awkwardness, tripping, or stumbling when walking or running; this is often marked by walking with a "dropped foot" that drags gently on the ground. If the arms are affected first, they may experience difficulty with tasks requiring manual dexterity, such as buttoning a shirt, writing, or turning a key in a lock.
In bulbar-onset ALS, the first symptoms are difficulty speaking or swallowing. Speech may become slurred, nasal in character, or quieter. There may be difficulty with swallowing and loss of tongue mobility. A smaller proportion of people experience "respiratory-onset" ALS, where the intercostal muscles that support breathing are affected first.
Over time, people experience increasing difficulty moving, swallowing (dysphagia), and speaking or forming words (dysarthria). Symptoms of upper motor neuron involvement include tight and stiff muscles (spasticity) and exaggerated reflexes (hyperreflexia), including an overactive gag reflex. While the disease does not cause pain directly, pain is a symptom experienced by most people with ALS caused by reduced mobility. Symptoms of lower motor neuron degeneration include muscle weakness and atrophy, muscle cramps, and fleeting twitches of muscles that can be seen under the skin (fasciculations).
Progression
Although the initial symptoms and rate of progression vary from person to person, the disease usually spreads to unaffected regions, and the affected regions become more severely affected. Most people eventually are not able to walk or use their hands and arms, lose the ability to speak and swallow food and their own saliva, and begin to lose the ability to cough and to breathe on their own.
The rate of progression can be measured using the ALS Functional Rating Scale - Revised (ALSFRS-R), a 12-item instrument survey administered as a clinical interview or self-reported questionnaire that produces a score between 48 (normal function) and 0 (severe disability). The ALSFRS-R is the most frequently used outcome measure in clinical trials and is used by doctors to track disease progression. Though the degree of variability is high and a small percentage of people have a much slower disorder, on average, people with ALS lose about 1 ALSFRS-r point per month. Brief periods of stabilization ("plateaus") and even small reversals in ALSFRS-r score are not uncommon, due to the fact the tool is subjective, can be affected by medication, and different forms of compensation for changes in function. However it is rare (<1%) for these improvements to be large (i.e. greater than 4 ALSFRS-r points) or sustained (i.e. greater than 12 months). A survey-based study among clinicians showed that they rated a 20% change in the slope of the ALSFRS-R as being clinically meaningful, which is the most common threshold used to determine whether a new treatment is working in clinical trials.
Late stage disease management
Difficulties with chewing and swallowing make eating very difficult and increase the risk of choking or of aspirating food into the lungs. In later stages of the disorder, aspiration pneumonia can develop, and maintaining a healthy weight can become a significant problem that may require the insertion of a feeding tube. As the diaphragm and intercostal muscles of the rib cage that support breathing weaken, measures of lung function such as vital capacity and inspiratory pressure diminish. In respiratory-onset ALS, this may occur before significant limb weakness is apparent. Individuals affected by the disorder may ultimately lose the ability to initiate and control all voluntary movement, known as locked-in syndrome. Bladder and bowel function is usually spared, meaning urinary and fecal incontinence is uncommon, although trouble getting to the toilet can lead to difficulties. The extraocular muscles responsible for eye movement are usually spared, meaning that the use of eye tracking technology to support augmentative communication is often feasible, albeit slow, and needs may change over time.
Prognosis, staging, and survival
Although respiratory support using non-invasive ventilation can ease problems with breathing and prolong survival, it does not affect the progression of ALS. Most people with ALS die between two and four years after the diagnosis. Around half of people with ALS die within 30 months of their symptoms beginning, and about 20% of people with ALS live between five and ten years after symptoms begin. About 10% of people with ALS survive for 10 years or longer after onset.
The most common cause of death among people with ALS is respiratory failure, often accelerated by pneumonia. Most ALS patients die at home after a period of worsening difficulty breathing, a decline in their nutritional status, or a rapid worsening of symptoms. Sudden death or acute respiratory distress are uncommon. Access to palliative care is recommended from an early stage to explore options, ensure psychosocial support for the patient and caregivers, and to discuss advance healthcare directives.
As with cancer staging, ALS has staging systems numbered between 1 and 4 that are used for research purposes in clinical trials. Two very similar staging systems emerged around a similar time, the King's staging system and Milano-Torino (MiToS) functional staging.
| Stage 0 | Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 | |
|---|---|---|---|---|---|---|
| King's Staging System | Not used | Symptom onset, involvement of the first region | 2A: Diagnosis
2B: Involvement of the second region |
Involvement of the third region | 4A: Need for a feeding tube
4B: Need for non-invasive ventilation |
Not used |
| Median time to King's stage | N/A | 13.5 months | 17.7 months | 23.3 months | 4A: 17.7 months
4B: 30.3 months |
N/A |
| ALS-MiToS Staging System | No loss of a functional domain | Loss of 1 domain | Loss of 2 domains | Loss of 3 domains | Loss of 4 domains | Death |
| Probability of death at each MiToS stage | 7% | 26% | 33% | 33% | 86% |
Providing individual patients with a precise prognosis is not currently possible, though research is underway to provide statistical models on the basis of prognostic factors including age at onset, progression rate, site of onset, and presence of frontotemporal dementia. Those with a bulbar onset have a worse prognosis than limb-onset ALS; a population-based study found that bulbar-onset ALS patients had a median survival of 2.0 years and a 10-year survival rate of 3%, while limb-onset ALS patients had a median survival of 2.6 years and a 10-year survival rate of 13%. Those with respiratory-onset ALS had a shorter median survival of 1.4 years and 0% survival at 10 years. While astrophysicist Stephen Hawking lived for 55 more years following his diagnosis, his was an unusual case.
Cognitive and behavioral symptoms
Cognitive or behavioral dysfunction is present in 30–50% of individuals with ALS. Around half of people with ALS will experience mild changes in cognition and behavior, and 10–15% will show signs of frontotemporal dementia (FTD). Most people with ALS who have normal cognition at the time of diagnosis have preserved cognition throughout the course of their disease; the development of cognitive impairment in those with normal cognition at baseline is associated with a worse prognosis.Repeating phrases or gestures, apathy, and loss of inhibition are frequently reported behavioral features of ALS.Language dysfunction, executive dysfunction, and troubles with social cognition and verbal memory are the most commonly reported cognitive symptoms in ALS; a meta-analysis found no relationship between dysfunction and disease severity. However, cognitive and behavioral dysfunctions have been found to correlate with reduced survival in people with ALS and increased caregiver burden; this may be due in part to deficits in social cognition. About half the people who have ALS experience emotional lability, in which they cry, smile, yawn, or laugh for no reason or when they are feeling the opposite emotion to that being expressed; it is more common in those with bulbar-onset ALS.
Cause
Nobody knows what causes ALS, hence it is described as an idiopathic disease. Though its exact cause is unknown, genetic and environmental factors are thought to be of roughly equal importance. The genetic factors are better understood than the environmental factors; no specific environmental factor has been definitively shown to cause ALS. A liability threshold model for ALS proposes that cellular damage accumulates over time due to genetic factors present at birth and exposure to environmental risks throughout life. ALS can strike at any age, but its likelihood increases with age. Most people who develop ALS are between the ages of 40 and 70, with an average age of 55 at the time of diagnosis. ALS is 20% more common in men than women, but the difference will disappear after age 70.
Genetics and genetic testing
ALS can be classified as being either genetic or sporadic, depending on whether or not there is a family history of the disease or whether an ALS-associated genetic mutation has been identified on testing. Genetic ALS is usually said to account for around 10% of all cases of ALS, though estimates range from 5% to 20%. In sporadic ALS, there is no family history of the disease. Sporadic ALS and genetic ALS appear identical clinically and pathologically.
There is considerable variation among clinicians on how to approach genetic testing in ALS, and only about half discuss the possibility of genetic inheritance with their patients, particularly if there is no discernible family history of the disease. Historically, genetic ALS was most commonly referred to as "familial" ALS and genetic counseling and testing only offered to those with a family history, though again there was a lack of consensus in the field as to what constituted a positive finding. The strictest definition was that a person with ALS must have two or more first-degree relatives (children, siblings, or parents) who also have been diagnosed with ALS. A less strict definition was that a person with ALS must have at least one first-degree or second-degree relative (grandparents, grandchildren, aunts, uncles, nephews, nieces or half-siblings) who also had a diagnosis of ALS. As a result of population-level genetic screening, we now know about 10% of people thought to have sporadic ALS also have mutations in genes that are known to cause ALS. The lack of family history may be caused by incomplete family history, older generations dying earlier of other causes than ALS, genetic non-paternity, or the occurrence of de novo mutations. There have been calls in the research community to routinely counsel and test all diagnosed ALS patients for genetic ALS, particularly as there is now a licensed gene therapy (tofersen) specifically targeted to carriers of SOD-1 ALS.
More than 40 genes have been associated with genetic ALS, of which four account for the majority of familial cases:C9orf72 (40% of genetic cases, 7% sporadic), SOD1 (12% of genetic cases, 1-2% sporadic), FUS (4% of genetic cases, 1% sporadic), and TARDBP (4% of genetic cases, 1% sporadic), with the remaining genes mostly accounting for fewer than 1% of genetic or sporadic cases. The genetics of familial ALS are better understood than the genetics of sporadic ALS; as of 2016, the known ALS genes explained about 70% of familial ALS and about 15% of sporadic ALS. Overall, first-degree relatives of an individual with ALS have a 1% risk of developing ALS. ALS has an oligogenic mode of inheritance, meaning that mutations in two or more genes are required to cause disease.
ALS and frontotemporal dementia (FTD) are now considered to be part of a common disease spectrum (ALS–FTD) because of genetic, clinical, and pathological similarities. Genetically, C9orf72 repeat expansions account for about 40% of genetic ALS and 25% of genetic FTD. Clinically, 50% of people with ALS have some cognitive or behavioral impairments and 5–15% have FTD, while 40% of people with FTD have some motor neuron symptoms and 12.5% have ALS. Pathologically, abnormal aggregations of TDP-43 protein are seen in up to 97% of ALS patients and up to 50% of FTD patients. Other genes known to cause ALS-FTD include C2orf72, CCNF, CHMP2B, CHCHD10, FUS, MATR3, SQSTM1, TARDBP, TBK1, TIA1, TUBA4A, UBQLN2, and VCP.
Environmental factors
Where no family history of the disease is present – around 90% of cases – no cause is known. Possible associations for which evidence is inconclusive include military service and smoking. Although studies on military history and ALS frequency are inconsistent, there is weak evidence for a positive correlation. Various proposed factors include exposure to environmental toxins (inferred from geographical deployment studies), as well as alcohol and tobacco use during military service.
A 2016 review of 16 meta-analyses concluded that there was convincing evidence for an association with chronic occupational exposure to lead; suggestive evidence for farming, exposure to heavy metals other than lead, beta-carotene intake, and head injury; and weak evidence for omega-3 fatty acid intake, exposure to extremely low frequency electromagnetic fields, pesticides, and serum uric acid.
In a 2017 study by the United States Centers for Disease Control and Prevention analyzing U.S. deaths from 1985 to 2011, occupations correlated with ALS deaths were white collar, such as in management, financial, architectural, computing, legal, and education jobs. Other potential risk factors remain unconfirmed, including chemical exposure, electromagnetic field exposure, occupation, physical trauma, and electric shock. There is a tentative association with exposure to various pesticides, including the organochlorine insecticides aldrin, dieldrin, DDT, and toxaphene.
Head injury
A 2015 review found that moderate to severe traumatic brain injury is a risk factor for ALS, but whether mild traumatic brain injury increases rates was unclear. A 2017 meta-analysis found an association between head injuries and ALS; however, this association disappeared when the authors considered the possibility of reverse causation, which is the idea that head injuries are an early symptom of undiagnosed ALS, rather than the cause of ALS.
Physical activity
A number of reviews prior to 2021 found no relationship between the amount of physical activity and the risk of developing ALS. A 2009 review found that the evidence for physical activity as a risk factor for ALS was limited, conflicting, and of insufficient quality to come to a firm conclusion. A 2014 review concluded that physical activity in general is not a risk factor for ALS, that football and American football are possibly associated with ALS, and that there was not enough evidence to say whether or not physically demanding occupations are associated with ALS. A 2016 review found the evidence inconclusive and noted that differences in study design make it difficult to compare studies, as they do not use the same measures of physical activity or the same diagnostic criteria for ALS. However, research published in 2021 suggested that there was a positive causal relationship between ALS and intense physical exercise in those with a risk genotype.
Sports
Both football and American football have been identified as risk factors for ALS in several studies, although this association is based on small numbers of ALS cases. A 2012 retrospective cohort study of 3,439 former NFL players found that their risk of dying from neurodegenerative causes was three times higher than the general US population, and their risk of dying from ALS or Alzheimer's disease was four times higher. However, this increased risk was calculated on the basis of two deaths from Alzheimer's disease and six deaths from ALS out of 334 deaths total in this cohort, meaning that this study does not definitively prove that playing American football is a risk factor for ALS. Some NFL players thought to have died from ALS may have actually had chronic traumatic encephalopathy (CTE), a neurodegenerative disorder associated with multiple head injuries that can present with symptoms that are very similar to ALS.
Football was identified as a possible risk factor for ALS in a retrospective cohort study of 24,000 Italian footballers who played between 1960 and 1996. There were 375 deaths in this group, including eight from ALS. Based on this information and the incidence of ALS, it was calculated that the football players were 11 times more likely to die from ALS than the general Italian population. However, this calculation has been criticized for relying on an inappropriately low number of expected cases of ALS in the cohort. When the lifetime risk of developing ALS was used to predict the number of expected cases, football players were no more likely to die of ALS than the general population.
Smoking
Smoking is possibly associated with ALS. A 2009 review concluded that smoking was an established risk factor for ALS. A 2010 systematic review and meta-analysis concluded that there was not a strong association between smoking and ALS, but that smoking might be associated with a higher risk of ALS in women. A 2011 meta-analysis concluded that smoking increases the risk of ALS versus never smoking. Among smokers, the younger they started smoking, the more likely they were to get ALS; however, neither the number of years smoked nor the number of cigarettes smoked per day affected their risk of developing ALS.
Pathophysiology
Neuropathology
The defining feature of ALS is the death of both upper motor neurons (located in the motor cortex of the brain) and lower motor neurons (located in the brainstem and spinal cord). In ALS with frontotemporal dementia, neurons throughout the frontal and temporal lobes of the brain die as well. The pathological hallmark of ALS is the presence of inclusion bodies (abnormal aggregations of protein) known as Bunina bodies in the cytoplasm of motor neurons. In about 97% of people with ALS, the main component of the inclusion bodies is TDP-43 protein; however, in those with SOD1 or FUS mutations, the main component of the inclusion bodies is SOD1 protein or FUS protein, respectively. The gross pathology of ALS, which are features of the disease that can be seen with the naked eye, including skeletal muscle atrophy, motor cortex atrophy, sclerosis of the corticospinal and corticobulbar tracts, thinning of the hypoglossal nerves (which control the tongue), and thinning of the anterior roots of the spinal cord. Aside from the death of motor neurons, two other characteristics common to most ALS variants are focal initial pathology, meaning that symptoms start in a single spinal cord region, and progressive continuous spread, meaning that symptoms spread to additional regions over time. Prion-like propagation of misfolded proteins from cell to cell may explain why ALS starts in one area and spreads to others. The glymphatic system may also be involved in the pathogenesis of ALS.
Biochemistry

It is still not fully understood why neurons die in ALS, but this neurodegeneration is thought to involve many different cellular and molecular processes. The genes known to be involved in ALS can be grouped into three general categories based on their normal function: protein degradation, the cytoskeleton, and RNA processing. Mutant SOD1 protein forms intracellular aggregations that inhibit protein degradation. Cytoplasmic aggregations of wild-type (normal) SOD1 protein are common in sporadic ALS. It is thought that misfolded mutant SOD1 can cause misfolding and aggregation of wild-type SOD1 in neighboring neurons in a prion-like manner. Other protein degradation genes that can cause ALS when mutated include VCP, OPTN, TBK1, and SQSTM1. Three genes implicated in ALS that are important for maintaining the cytoskeleton and for axonal transport include DCTN1, PFN1, and TUBA4A.
There are a number of ALS genes that encode for RNA-binding proteins. The first to be discovered was TDP-43 protein, a nuclear protein that aggregates in the cytoplasm of motor neurons in almost all cases of ALS; however, mutations in TARDBP, the gene that codes for TDP-43, are a rare cause of ALS.FUS codes for FUS, another RNA-binding protein with a similar function to TDP-43, which can cause ALS when mutated. It is thought that mutations in TARDBP and FUS increase the binding affinity of the low-complexity domain, causing their respective proteins to aggregate in the cytoplasm. Once these mutant RNA-binding proteins are misfolded and aggregated, they may be able to misfold normal proteins both within and between cells in a prion-like manner. This also leads to decreased levels of RNA-binding protein in the nucleus, which may mean that their target RNA transcripts do not undergo normal processing. Other RNA metabolism genes associated with ALS include ANG, SETX, and MATR3.
C9orf72 is the most commonly mutated gene in ALS and causes motor neuron death through a number of mechanisms. The pathogenic mutation is a hexanucleotide repeat expansion (a series of six nucleotides repeated over and over); people with up to 30 repeats are considered normal, while people with hundreds or thousands of repeats can have familial ALS, frontotemporal dementia, or sometimes sporadic ALS. The three mechanisms of disease associated with these C9orf72 repeats are deposition of RNA transcripts in the nucleus, translation of the RNA into toxic dipeptide repeat proteins in the cytoplasm, and decreased levels of the normal C9orf72 protein. Mitochondrial bioenergetic dysfunction leading to dysfunctional motor neuron axonal homeostasis (reduced axonal length and fast axonal transport of mitochondrial cargo) has been shown to occur in C9orf72-ALS using human induced pluripotent stem cell (iPSC) technologies coupled with CRISPR/Cas9 gene-editing, and human post-mortem spinal cord tissue examination.
Excitotoxicity, or nerve cell death caused by high levels of intracellular calcium due to excessive stimulation by the excitatory neurotransmitter glutamate, is a mechanism thought to be common to all forms of ALS. Motor neurons are more sensitive to excitotoxicity than other types of neurons because they have a lower calcium-buffering capacity and a type of glutamate receptor (the AMPA receptor) that is more permeable to calcium. In ALS, there are decreased levels of excitatory amino acid transporter 2 (EAAT2), which is the main transporter that removes glutamate from the synapse; this leads to increased synaptic glutamate levels and excitotoxicity. Riluzole, a drug that modestly prolongs survival in ALS, inhibits glutamate release from pre-synaptic neurons; however, it is unclear if this mechanism is responsible for its therapeutic effect.
Diagnosis

No single test can provide a definite diagnosis of ALS. Instead, the diagnosis of ALS is primarily based on the symptoms and signs the physician observes in the person and a series of tests to rule out other diseases. Physicians obtain the person's full medical history and usually conduct a neurologic examination at regular intervals to assess whether signs and symptoms such as muscle weakness, atrophy of muscles, hyperreflexia, Babinski's sign, and spasticity are worsening. A number of biomarkers are being studied for the condition, but so far are not in general medical use.
Diagnostic criteria

The diagnosis of ALS is based on the El Escorial Revised criteria and the Awaji criteria. The original El Escorial criteria had four levels of diagnostic certainty, based on how many of the four spinal cord regions were involved: bulbar, cervical, thoracic, and lumbar. Definite ALS was defined as upper motor neuron (UMN) and lower motor neuron (LMN) signs in three spinal cord regions, probable ALS as UMN and LMN signs in two regions, possible ALS as UMN and LMN signs in only one region, and suspected ALS as LMN signs only. The El Escorial Revised criteria, also known as the Airlie House criteria, dropped the "suspected ALS" category and added a "laboratory-supported probable ALS" category. The Awaji criteria give abnormal EMG tests the same weight as clinical signs of LMN dysfunction in making the diagnosis of ALS, thus making the "laboratory-supported probable ALS" category unnecessary. The only three categories in the Awaji criteria are definite ALS, probable ALS, and possible ALS.
The El Escorial Revised criteria are specific for ALS, which means that someone who meets the criteria is very likely to have ALS; however, they are not especially sensitive for ALS, which means that someone who does not meet the criteria can still have ALS. Their sensitivity is particularly poor in the early stages of ALS. The Awaji criteria have better sensitivity than the El Escorial Revised criteria, especially for bulbar-onset ALS. A 2012 meta-analysis found that the El Escorial Revised criteria had a sensitivity of 62.2%, while the Awaji criteria had a sensitivity of 81.1%; both sets of criteria had a specificity of about 98%. The El Escorial criteria were designed to standardize patient groups for clinical trials but are not as useful in clinical practice; possible ALS as described by the El Escorial criteria is almost always clinically ALS.
Differential diagnosis
Because symptoms of ALS can be similar to those of a wide variety of other, more treatable diseases or disorders, appropriate tests must be conducted to exclude the possibility of other conditions. One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in muscles. Certain EMG findings can support the diagnosis of ALS. Another common test measures nerve conduction velocity (NCV). Specific abnormalities in the NCV results may suggest, for example, that the person has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) rather than ALS. While a magnetic resonance imaging (MRI) is often normal in people with early-stage ALS, it can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, multiple sclerosis, a herniated disc in the neck, syringomyelia, or cervical spondylosis.
Based on the person's symptoms and findings from the examination and from these tests, the physician may order tests on blood and urine samples to eliminate the possibility of other diseases, as well as routine laboratory tests. In some cases, for example, if a physician suspects the person may have a myopathy rather than ALS, a muscle biopsy may be performed.
A number of infectious diseases can sometimes cause ALS-like symptoms, including human immunodeficiency virus (HIV), human T-lymphotropic virus (HTLV), Lyme disease, and syphilis. Neurological disorders such as multiple sclerosis, post-polio syndrome, multifocal motor neuropathy, CIDP, spinal muscular atrophy, and spinal and bulbar muscular atrophy can also mimic certain aspects of the disease and should be considered.
ALS must be differentiated from the "ALS mimic syndromes", which are unrelated disorders that may have a similar presentation and clinical features to ALS or its variants. Because the prognosis of ALS and closely related subtypes of motor neuron disease are generally poor, neurologists may carry out investigations to evaluate and exclude other diagnostic possibilities. Disorders of the neuromuscular junction, such as myasthenia gravis (MG) and Lambert–Eaton myasthenic syndrome, may also mimic ALS, although this rarely presents diagnostic difficulty over time.Benign fasciculation syndrome and cramp fasciculation syndrome may also, occasionally, mimic some of the early symptoms of ALS. Nonetheless, the absence of other neurological features that develop inexorably with ALS means that, over time, the distinction will not present any difficulty to the experienced neurologist; where doubt remains, EMG may be helpful.
Most cases of ALS, however, are correctly diagnosed, with the error rate of diagnosis in large ALS clinics being less than 10%. One study examined 190 people who met the MND/ALS diagnostic criteria, complemented with laboratory research in compliance with both research protocols and regular monitoring. Thirty of these people (16%) had their diagnosis completely changed during the clinical observation development period. In the same study, three people had a false negative diagnosis of MG, which can mimic ALS and other neurological disorders, leading to a delay in diagnosis and treatment. MG is eminently treatable; ALS is not.
Management
There is no cure for ALS. Management focuses on treating symptoms and providing supportive care, with the goal of improving quality of life and prolonging survival. This care is best provided by multidisciplinary teams of healthcare professionals; attending a multidisciplinary ALS clinic is associated with longer survival, fewer hospitalizations, and improved quality of life. Riluzole prolongs survival by about 2–3 months.Edaravone slows functional decline slightly in a small number of people with ALS; it is expensive and must be administered by daily IV infusions that may decrease quality of life. Other medications may be used to manage other symptoms.
Non-invasive ventilation (NIV) is the main treatment for respiratory failure in ALS. In people with normal bulbar function, it prolongs survival by about seven months and improves quality of life. One study found that NIV is ineffective for people with poor bulbar function while another suggested that it may provide a modest survival benefit. Many people with ALS have difficulty tolerating NIV. Invasive ventilation is an option for people with advanced ALS when NIV is not enough to manage their symptoms. While invasive ventilation prolongs survival, disease progression and functional decline continue. It may decrease the quality of life of people with ALS or their caregivers. Invasive ventilation is more commonly used in Japan than in North America or Europe.
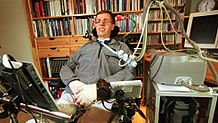
Physical therapy can promote functional independence through an aerobic, range of motion, and stretching exercises. Occupational therapy can assist with activities of daily living through adaptive equipment. Speech therapy can assist people with ALS who have difficulty speaking. Preventing weight loss and malnutrition in people with ALS improves both survival and quality of life. Initially, difficulty swallowing (dysphagia) can be managed by dietary changes and swallowing techniques. A feeding tube should be considered if someone with ALS loses 5% or more of their body weight or if they cannot safely swallow food and water. The feeding tube is usually inserted by percutaneous endoscopic gastrostomy (PEG). There is weak evidence that PEG tubes improve survival. PEG insertion is usually performed with the intent of improving quality of life.
Palliative care should begin shortly after someone is diagnosed with ALS. Discussion of end-of-life issues gives people with ALS time to reflect on their preferences for end-of-life care and can help avoid unwanted interventions or procedures. Hospice care can improve symptom management at the end of life and increases the likelihood of a peaceful death. In the final days of life, opioids can be used to treat pain and dyspnea, while benzodiazepines can be used to treat anxiety.
Medications
Disease-slowing treatments

Riluzole has been found to modestly prolong survival by about 2–3 months. It may have a greater survival benefit for those with bulbar-onset ALS. It may work by decreasing release of the excitatory neurotransmitter glutamate from pre-synaptic neurons. The most common side effects are nausea and a lack of energy (asthenia). People with ALS should begin treatment with riluzole as soon as possible following their diagnosis. Riluzole is available as a tablet, liquid, or dissolvable oral film.
Edaravone has been shown to modestly slow the decline in function in a small group of people with early-stage ALS. It may work by protecting motor neurons from oxidative stress. The most common side effects are bruising and gait disturbance. Edaravone is available as an intravenous infusion or as an oral suspension.
AMX0035, a combination of sodium phenylbutyrate and taurursodiol, was shown to prolong the survival of patients by an average of six months.
Tofersen (Qalsody) is an antisense oligonucleotide that was approved for medical use in the United States in April 2023, for the treatment of SOD1-associated ALS. In a study of 108 patients with SOD1-associated ALS there was a non-significant trend towards a slowing of progression, as well as a 35%+50% reduction in the putative ALS biomarker neurofilament light chain, thought to indicate neuronal damage. A follow-up study and open-label extension suggested that earlier treatment initiation had a beneficial effect on slowing disease progression. Tofersen is available as an injection into the spinal cord.
Symptomatic treatments
Other medications may be used to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and phlegm.Gabapentin, pregabalin, and tricyclic antidepressants (e.g., amitriptyline) can be used for neuropathic pain, while nonsteroidal anti-inflammatory drugs (NSAIDs), acetaminophen, and opioids can be used for nociceptive pain.
Depression can be treated with selective serotonin reuptake inhibitors (SSRIs) or tricyclic antidepressants, while benzodiazepines can be used for anxiety. There are no medications to treat cognitive impairment/frontotemporal dementia (FTD); however, SSRIs and antipsychotics can help treat some of the symptoms of FTD.Baclofen and tizanidine are the most commonly used oral drugs for treating spasticity; an intrathecal baclofen pump can be used for severe spasticity.Atropine, scopolamine, amitriptyline or glycopyrrolate may be prescribed when people with ALS begin having trouble swallowing their saliva (sialorrhea).
A 2017 review concluded that mexiletine is safe and effective for treating cramps in ALS based on a randomized controlled trial from 2016.
Breathing support
Non-invasive ventilation

Non-invasive ventilation (NIV) is the primary treatment for respiratory failure in ALS and was the first treatment shown to improve both survival and quality of life. NIV uses a face or nasal mask connected to a ventilator that provides intermittent positive pressure to support breathing. Continuous positive pressure is not recommended for people with ALS because it makes breathing more difficult. Initially, NIV is used only at night because the first sign of respiratory failure is decreased gas exchange (hypoventilation) during sleep; symptoms associated with this nocturnal hypoventilation include interrupted sleep, anxiety, morning headaches, and daytime fatigue. As the disease progresses, people with ALS develop shortness of breath when lying down, during physical activity or talking, and eventually at rest. Other symptoms include poor concentration, poor memory, confusion, respiratory tract infections, and a weak cough. Respiratory failure is the most common cause of death in ALS.
It is important to monitor the respiratory function of people with ALS every three months because beginning NIV soon after the start of respiratory symptoms is associated with increased survival. This involves asking the person with ALS if they have any respiratory symptoms and measuring their respiratory function. The most commonly used measurement is upright forced vital capacity (FVC), but it is a poor detector of early respiratory failure and is not a good choice for those with bulbar symptoms, as they have difficulty maintaining a tight seal around the mouthpiece. Measuring FVC while the person is lying on their back (supine FVC) is a more accurate measure of diaphragm weakness than upright FVC. Sniff nasal inspiratory pressure (SNIP) is a rapid, convenient test of diaphragm strength that is not affected by bulbar muscle weakness. If someone with ALS has signs and symptoms of respiratory failure, they should undergo daytime blood gas analysis to look for hypoxemia (low oxygen in the blood) and hypercapnia (too much carbon dioxide in the blood). If their daytime blood gas analysis is normal, they should then have nocturnal pulse oximetry to look for hypoxemia during sleep.
Non-invasive ventilation prolongs survival longer than riluzole. A 2006 randomized controlled trial found that NIV prolongs survival by about 48 days and improves the quality of life; however, it also found that some people with ALS benefit more from this intervention than others. For those with normal or only moderately impaired bulbar function, NIV prolongs survival by about seven months and significantly improves the quality of life. For those with poor bulbar function, NIV neither prolongs survival nor improves the quality of life, though it does improve some sleep-related symptoms. Despite the clear benefits of NIV, about 25–30% of all people with ALS are unable to tolerate it, especially those with cognitive impairment or bulbar dysfunction. Results from a large 2015 cohort study suggest that NIV may prolong survival in those with bulbar weakness, so NIV should be offered to all people with ALS, even if it is likely that they will have difficulty tolerating it.
Invasive ventilation
Invasive ventilation bypasses the nose and mouth (the upper airways) by making a cut in the trachea (tracheostomy) and inserting a tube connected to a ventilator. It is an option for people with advanced ALS whose respiratory symptoms are poorly managed despite continuous NIV use. While invasive ventilation prolongs survival, especially for those younger than 60, it does not treat the underlying neurodegenerative process. The person with ALS will continue to lose motor function, making communication increasingly difficult and sometimes leading to locked-in syndrome, in which they are completely paralyzed except for their eye muscles. About half of the people with ALS who choose to undergo invasive ventilation report a decrease in their quality of life but most still consider it to be satisfactory. However, invasive ventilation imposes a heavy burden on caregivers and may decrease their quality of life. Attitudes toward invasive ventilation vary from country to country; about 30% of people with ALS in Japan choose invasive ventilation, versus less than 5% in North America and Europe.
Therapy

Physical therapy plays a large role in rehabilitation for individuals with ALS. Specifically, physical, occupational, and speech therapists can set goals and promote benefits for individuals with ALS by delaying loss of strength, maintaining endurance, limiting pain, improving speech and swallowing, preventing complications, and promoting functional independence.
Occupational therapy and special equipment such as assistive technology can also enhance people's independence and safety throughout the course of ALS. Gentle, low-impact aerobic exercise such as performing activities of daily living, walking, swimming, and stationary bicycling can strengthen unaffected muscles, improve cardiovascular health, and help people fight fatigue and depression. Range of motion and stretching exercises can help prevent painful spasticity and shortening (contracture) of muscles. Physical and occupational therapists can recommend exercises that provide these benefits without overworking muscles because muscle exhaustion can lead to a worsening of symptoms associated with ALS, rather than providing help to people with ALS. They can suggest devices such as ramps, braces, walkers, bathroom equipment (shower chairs, toilet risers, etc.), and wheelchairs that help people remain mobile. Occupational therapists can provide or recommend equipment and adaptations to enable ALS people to retain as much safety and independence in activities of daily living as possible. Since respiratory insufficiency is the primary cause of mortality, physical therapists can help improve respiratory outcomes in people with ALS by implementing pulmonary physical therapy. This includes inspiratory muscle training, lung volume recruitment training, and manual assisted cough therapy aimed at increasing respiratory muscle strength as well as increasing survival rates.
People with ALS who have difficulty speaking or swallowing may benefit from working with a speech-language pathologist. These health professionals can teach people adaptive strategies such as techniques to help them speak louder and more clearly. As ALS progresses, speech-language pathologists can recommend the use of augmentative and alternative communication such as voice amplifiers, speech-generating devices (or voice output communication devices) or low-tech communication techniques such as head-mounted laser pointers, alphabet boards or yes/no signals.
In a study published in 2022, a completely locked-in ALS patient was fitted with two 64-bit brain implant microarrays in his motor cortex in 2020. Using audible feedback from his own intentional neural firing rates, he was able to communicate letters to form phrases. This is the first example of communication at length with a fully locked-in ALS patient.
Nutrition

Preventing weight loss and malnutrition in people with ALS improves both survival and quality of life. Weight loss in ALS is caused by muscle wasting due to motor neuron death increased resting energy expenditure, and decreased food intake. Difficulty swallowing (dysphagia) develops in about 85% of people with ALS at some point over the course of their disease and is a major cause of decreased food intake, leading to malnutrition and weight loss. It is important to regularly assess the weight and swallowing ability of people with ALS. Initially, dysphagia may be managed by dietary changes and modified swallowing techniques. Difficulty swallowing liquids usually develops first and can be managed by switching to thicker liquids like fruit nectar or smoothies, or by adding fluid thickeners to thin fluids like water and coffee. People with ALS should eat soft, moist foods, which tend to be easier to swallow than dry, crumbly, or chewy foods. They should also be instructed on proper head posture during swallowing, which can make swallowing easier. There is tentative evidence that high-calorie diets may prevent further weight loss and improve survival.
A feeding tube should be considered if someone with ALS loses 5% or more of their body weight or if they cannot safely swallow food and water. This can take the form of a gastrostomy tube, in which a tube is placed through the wall of the abdomen into the stomach, or a nasogastric tube, in which a tube is placed through the nose and down the esophagus into the stomach. A gastrostomy tube is more appropriate for long-term use than a nasogastric tube, which is uncomfortable and can cause esophageal ulcers. The feeding tube is usually inserted by a percutaneous endoscopic gastrostomy procedure (PEG).
There is weak evidence that PEG tubes improve survival. PEG insertion is usually performed with the intent of improving quality of life by sustaining nutrition and medication intake. This reduces the risk of weight loss and dehydration and can decrease anxiety from extended mealtimes and decreased oral food intake.
End-of-life care
Palliative care, which relieves symptoms and improves the quality of life without treating the underlying disease, should begin shortly after someone is diagnosed with ALS. Early discussion of end-of-life issues gives people with ALS time to reflect on their preferences for end-of-life care and can help avoid unwanted interventions or procedures. Once they have been fully informed about all aspects of various life-prolonging measures, they can fill out advance directives indicating their attitude toward noninvasive ventilation, invasive ventilation, and feeding tubes. Late in the disease course, difficulty speaking due to muscle weakness (dysarthria) and cognitive dysfunction may impair their ability to communicate their wishes regarding care. Continued failure to solicit the preferences of the person with ALS may lead to unplanned and potentially unwanted emergency interventions, such as invasive ventilation. If people with ALS or their family members are reluctant to discuss end-of-life issues, it may be useful to use the introduction of gastrostomy or noninvasive ventilation as an opportunity to bring up the subject.
Hospice care, or palliative care at the end of life, is especially important in ALS because it helps to optimize the management of symptoms and increases the likelihood of a peaceful death. It is unclear exactly when the end-of-life phase begins in ALS, but it is associated with significant difficulty moving, communicating, and, in some cases, thinking. Although many people with ALS fear choking to death (suffocating), they can be reassured that this occurs rarely, about 0–3% of the time. About 90% of people with ALS die peacefully. In the final days of life, opioids can be used to treat pain and dyspnea, while benzodiazepines can be used to treat anxiety.
Epidemiology
ALS is the most common motor neuron disease in adults and the third most common neurodegenerative disease after Alzheimer's disease and Parkinson's disease. Worldwide the number of people who develop ALS yearly is estimated to be 1.9 people per 100,000 per year, while the number of people who have ALS at any given time is estimated to be about 4.5 people per 100,000. In Europe, the number of new cases a year is about 2.6 people per 100,000, while the number affected is 7–9 people per 100,000. The lifetime risk of developing ALS is 1:350 for European men and 1:400 for European women. Men have a higher risk mainly because spinal-onset ALS is more common in men than women. The number of those with ALS in the United States in 2015 was 5.2 people per 100,000, and was higher in whites, males, and people over 60 years old. The number of new cases is about 0.8 people per 100,000 per year in east Asia and about 0.7 people per 100,000 per year in south Asia. About 80% of ALS epidemiology studies have been conducted in Europe and the United States, mostly in people of northern European descent. There is not enough information to determine the rates of ALS in much of the world, including Africa, parts of Asia, India, Russia, and South America. There are several geographic clusters in the Western Pacific where the prevalence of ALS was reported to be 50–100 times higher than the rest of the world, including Guam, the Kii Peninsula of Japan, and Western New Guinea. The incidence in these areas has decreased since the 1960s; the cause remains unknown.

People of all races and ethnic backgrounds may be affected by ALS, but it is more common in whites than in Africans, Asians, or Hispanics. In the United States in 2015, the prevalence of ALS in whites was 5.4 people per 100,000, while the prevalence in blacks was 2.3 people per 100,000. The Midwest had the highest prevalence of the four US Census regions with 5.5 people per 100,000, followed by the Northeast (5.1), the South (4.7), and the West (4.4). The Midwest and Northeast likely had a higher prevalence of ALS because they have a higher proportion of whites than the South and West. Ethnically mixed populations may be at a lower risk of developing ALS; a study in Cuba found that people of mixed ancestry were less likely to die from ALS than whites or blacks. There are also differences in the genetics of ALS between different ethnic groups; the most common ALS gene in Europe is C9orf72, followed by SOD1, TARDBP, and FUS, while the most common ALS gene in Asia is SOD1, followed by FUS, C9orf72, and TARDBP.
ALS can affect people at any age, but the peak incidence is between 50 and 75 years and decreases dramatically after 80 years. The reason for the decreased incidence in the elderly is unclear. One thought is that people who survive into their 80s may not be genetically susceptible to developing ALS; alternatively, ALS in the elderly might go undiagnosed because of comorbidities (other diseases they have), difficulty seeing a neurologist, or dying quickly from an aggressive form of ALS. In the United States in 2015, the lowest prevalence was in the 18–39 age group, while the highest prevalence was in the 70–79 age group. Sporadic ALS usually starts around the ages of 58 to 63 years, while familial ALS starts earlier, usually around 47 to 52 years. The number of ALS cases worldwide is projected to increase from 222,801 in 2015 to 376,674 in 2040, an increase of 69%. This will largely be due to the aging of the world's population, especially in developing countries.
History


Descriptions of the disease date back to at least 1824 by Charles Bell. In 1850, François-Amilcar Aran was the first to describe a disorder he named "progressive muscular atrophy", a form of ALS in which only the lower motor neurons are affected. In 1869, the connection between the symptoms and the underlying neurological problems were first described by Jean-Martin Charcot, who initially introduced the term amyotrophic lateral sclerosis in his 1874 paper. Flail arm syndrome, a regional variant of ALS, was first described by Alfred Vulpian in 1886. Flail leg syndrome, another regional variant of ALS, was first described by Pierre Marie and his student Patrikios in 1918.
Diagnostic criteria
In the 1950s, electrodiagnostic testing (EMG and NCV) began to be used to evaluate clinically suspected ALS. In 1969 Edward H. Lambert published the first EMG/NCS diagnostic criteria for ALS, consisting of four findings he considered to strongly support the diagnosis. In 1990, the World Federation of Neurology (WFN) held a meeting at El Escorial, Spain, to come up with precise diagnostic criteria for ALS to help standardize clinical trials; the resulting "El Escorial" criteria were published in 1994. In 1998, the WFN held another meeting to revise the criteria at Airlie House in Warrenton, Virginia; the resulting "Airlie House" or "El Escorial Revised" criteria were published in 2000. In 2006, a meeting was held on Awaji Island in Japan to discuss how to use EMG and NCV tests to help diagnose ALS earlier; the resulting "Awaji" criteria were published in 2008.
Name
Amyotrophic comes from Greek: a- means "no", myo- (from mûs) refers to "muscle", and trophḗ means "nourishment". Therefore, amyotrophy means "muscle malnourishment" or the wasting of muscle tissue.Lateral identifies the areas in a person's spinal cord where the affected motor neurons that control muscle are located. Sclerosis means "scarring" or "hardening" and refers to the death of the motor neurons in the spinal cord.
ALS is sometimes referred to as Charcot's disease (not to be confused with Charcot–Marie–Tooth disease or Charcot joint disease), because Jean-Martin Charcot was the first to connect the clinical symptoms with the pathology seen at autopsy. The British neurologist Russell Brain coined the term motor neurone disease in 1933 to reflect his belief that ALS, progressive bulbar palsy, and progressive muscular atrophy were all different forms of the same disease. In some countries, especially the United States, ALS is called Lou Gehrig's disease after the American baseball player Lou Gehrig, who developed ALS in 1938.
In the United States and continental Europe, the term ALS (as well as Lou Gehrig's disease in the US) refers to all forms of the disease, including "classical" ALS, progressive bulbar palsy, progressive muscular atrophy, and primary lateral sclerosis. In the United Kingdom and Australia, the term motor neurone disease refers to all forms of the disease while ALS only refers to "classical" ALS, meaning the form with both upper and lower motor neuron involvement.
Society and culture
People with ALS have been featured in high-profile works such as the memoir Tuesdays with Morrie and the critically acclaimed motion picture The Theory of Everything.
In August 2014 the "ALS Ice Bucket Challenge" went viral online. Contestants filled a bucket full of ice and water, stated who nominated them to do the challenge, and nominated three other individuals. The contestants then poured the buckets of ice and water onto themselves. Many contestants then donated to ALS research at the ALS Association, the ALS Therapy Development Institute, ALS Society of Canada, or Motor Neurone Disease Association in the UK.
External links
- ALS Association Official Website
- ALS Therapy Development Institute
- International Alliance of ALS/MND Associations
- International Symposium on ALS/MND