
| Alexander disease | |
|---|---|
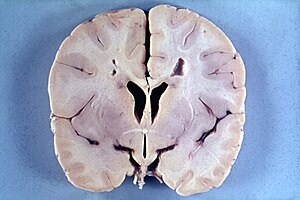 | |
| Brain of a 4-year-old boy with Alexander disease showing macroencephaly and periventricular leukomalacia (note brownish discoloration around the cerebral ventricles) | |
| Specialty |
Endocrinology, neurology |
Alexander disease is a very rare autosomal dominant leukodystrophy, which are neurological conditions caused by anomalies in the myelin which protects nerve fibers in the brain. The most common type is the infantile form that usually begins during the first two years of life. Symptoms include mental and physical developmental delays, followed by the loss of developmental milestones, an abnormal increase in head size and seizures. The juvenile form of Alexander disease has an onset between the ages of 2 and 13 years. These children may have excessive vomiting, difficulty swallowing and speaking, poor coordination, and loss of motor control. Adult-onset forms of Alexander disease are less common. The symptoms sometimes mimic those of Parkinson’s disease or multiple sclerosis, or may present primarily as a psychiatric disorder.
According to the National Institute of Neurological Disorders and Stroke, the destruction of white matter is accompanied by the formation of Rosenthal fibers—abnormal clumps of protein that accumulate in astrocytes in the brain.
The disease occurs in both males and females, and no ethnic, racial, geographic or cultural/economic differences are seen in its distribution. Alexander disease is a progressive and often fatal disease.
Presentation
Delays in development of some physical, psychological and behavioral skills; progressive enlargement of the head (macrocephaly), seizures, spasticity, and in some cases also hydrocephalus, idiopathic intracranial hypertension, and dementia.
In cases of early-onset or neonatal Alexander disease, symptoms include seizures, fluid buildup in the brain, high protein levels in cerebrospinal fluid, and severe motor and intellectual impairment. In cases of type I Alexander disease, where the condition appears before age 4, symptoms include seizures, enlarged brain and head, stiffness in the limbs, delayed intellectual and physical development, recurrent vomiting, and difficulties with gaining weight. In cases of type II Alexander disease, where the condition appears after the age of 4, symptoms include speech problems, difficulty swallowing, poor coordination, scoliosis, recurrent vomiting, and difficulties with gaining weight.
Cause
Alexander disease is a genetic disorder affecting the midbrain and cerebellum of the central nervous system. It is caused by mutations in the gene for glial fibrillary acidic protein (GFAP) that maps to chromosome 17q21. It is inherited in an autosomal dominant manner, such that the child of a parent with the disease has a 50% chance of inheriting the condition, if the parent is heterozygotic. However, most cases arise de novo as the result of sporadic mutations.
Alexander disease belongs to leukodystrophies, a group of diseases that affect the growth or development of the myelin sheath. The destruction of white matter in the brain is accompanied by the formation of fibrous, eosinophilic deposits known as Rosenthal fibers. Rosenthal fibers appear not to be present in healthy people, but occur in specific diseases, like some forms of cancer, Alzheimer’s, Parkinson’s, Huntington’s, and ALS. The Rosenthal fibers found in Alexander disease do not share the distribution or concentration of other diseases and disorders.
Pathology
Alexander disease causes the gradual loss of bodily functions and the ability to talk. It also causes an overload of long-chain fatty acids in the brain, which destroy the myelin sheath. The cause of Alexander disease is a mutation in the gene encoding GFAP.
A CT scan shows:
- Decreased density of white matter
- Frontal lobe predominance
- Dilated lateral ventricles may present
Diagnosis
Detecting the signs of Alexander disease is possible with magnetic resonance imaging (MRI), which looks for specific changes in the brain that may be tell-tale signs for the disease. It is even possible to detect adult-onset Alexander disease with MRI. Alexander disease may also be revealed by genetic testing for its known cause. A rough diagnosis may also be made through revealing of clinical symptoms, including enlarged head size, along with radiological studies, and negative tests for other leukodystrophies.
Treatment
No cure or standard procedure for treatment is known, although a University of Wisconsin study shows promise with gene editing of the astrocytes. A phase III clinical trial of an antisense therapy, sponsored by Ionis Pharmaceuticals, began in 2021. A bone marrow transplant has been attempted on a child, but it made no improvement. Hydrocephalus may be seen in younger patients and can be relieved with surgery or by implanting a shunt to relieve pressure.
Prognosis
The prognosis is generally poor. With early onset, death usually occurs within 10 years from the onset of symptoms. Individuals with the infantile form usually die before the age of seven. Usually, the later the disease occurs, the slower its course.
Prevalence
Its occurrence is very rare. The infantile form occurs from birth to two years of age. The average duration of the infantile form is usually about three years. Onset of the juvenile form presents between 2 and 12 years of age. Duration of this form is in most cases about six years. The adult form occurs after 12 years. In younger patients, seizures, megalencephaly, developmental delay, and spasticity are usually present. Neonatal onset is also reported. Onset in adults is least frequent. In older patients, bulbar or pseudobulbar symptoms and spasticity predominate. Symptoms of the adult form may also resemble multiple sclerosis. No more than 500 cases have been reported.