
| Amyloid beta peptide (beta-APP) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
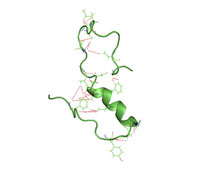 A partially folded structure of amyloid beta(1 40) in an aqueous environment (pdb 2lfm)
| |||||||||
| Identifiers | |||||||||
| Symbol | APP | ||||||||
| Pfam | PF03494 | ||||||||
| InterPro | IPR013803 | ||||||||
| SCOP2 | 2lfm / SCOPe / SUPFAM | ||||||||
| TCDB | 1.C.50 | ||||||||
| OPM superfamily | 304 | ||||||||
| OPM protein | 2y3k | ||||||||
| Membranome | 45 | ||||||||
| |||||||||
| amyloid beta (A4) precursor protein (peptidase nexin-II, Alzheimer disease) | |||||||
|---|---|---|---|---|---|---|---|
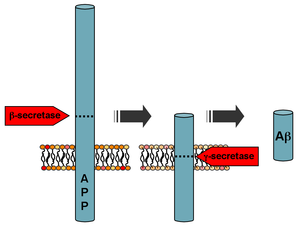 Processing of the amyloid precursor protein
| |||||||
| Identifiers | |||||||
| Symbol | APP | ||||||
| Alt. symbols | AD1 | ||||||
| NCBI gene | 351 | ||||||
| HGNC | 620 | ||||||
| OMIM | 104760 | ||||||
| RefSeq | NM_000484 | ||||||
| UniProt | P05067 | ||||||
| Other data | |||||||
| Locus | Chr. 21 q21.2 | ||||||
| |||||||
Amyloid beta (Aβ or Abeta) denotes peptides of 36–43 amino acids that are the main component of the amyloid plaques found in the brains of people with Alzheimer's disease. The peptides derive from the amyloid-beta precursor protein (APP), which is cleaved by beta secretase and gamma secretase to yield Aβ in a cholesterol-dependent process and substrate presentation. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers (known as "seeds") can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The oligomers are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.
A study has suggested that APP and its amyloid potential is of ancient origins, dating as far back as early deuterostomes.
Normal function
The normal function of Aβ is not well understood. Though some animal studies have shown that the absence of Aβ does not lead to any obvious loss of physiological function, several potential activities have been discovered for Aβ, including activation of kinase enzymes, protection against oxidative stress, regulation of cholesterol transport, functioning as a transcription factor, and anti-microbial activity (potentially associated with Aβ's pro-inflammatory activity).
The glymphatic system clears metabolic waste from the mammalian brain, and in particular amyloid beta. Indeed, a number of proteases have been implicated by both genetic and biochemical studies as being responsible for the recognition and degradation of amyloid beta; these include insulin degrading enzyme and presequence protease. The rate of removal is significantly increased during sleep. However, the significance of the glymphatic system in Aβ clearance in Alzheimer's disease is unknown.
Disease associations
Aβ is the main component of amyloid plaques, extracellular deposits found in the brains of people with Alzheimer's disease. Aβ can also form the deposits that line cerebral blood vessels in cerebral amyloid angiopathy. The plaques are composed of a tangle of Aβ oligomers and regularly ordered aggregates called amyloid fibrils, a protein fold shared by other peptides such as the prions associated with protein misfolding diseases.
Alzheimer's disease
Research suggests that soluble oligomeric forms of the peptide may be causative agents in the development of Alzheimer's disease. It is generally believed that Aβ oligomers are the most toxic. The ion channel hypothesis postulates that oligomers of soluble, non-fibrillar Aβ form membrane ion channels allowing the unregulated calcium influx into neurons that underlies disrupted calcium ion homeostasis and apoptosis seen in Alzheimer's disease. Computational studies have demonstrated that also Aβ peptides embedded into the membrane as monomers with predominant helical configuration, can oligomerize and eventually form channels whose stability and conformation are sensitively correlated to the concomitant presence and arrangement of cholesterol. A number of genetic, cell biology, biochemical and animal studies using experimental models support the concept that Aβ plays a central role in the development of Alzheimer's disease pathology.
Brain Aβ is elevated in people with sporadic Alzheimer's disease. Aβ is the main constituent of brain parenchymal and vascular amyloid; it contributes to cerebrovascular lesions and is neurotoxic. It is unresolved how Aβ accumulates in the central nervous system and subsequently initiates the disease of cells. Some researchers have found that the Aβ oligomers induce some of the symptoms of Alzheimer's disease by competing with insulin for binding sites on the insulin receptor, thus impairing glucose metabolism in the brain. Significant efforts have been focused on the mechanisms responsible for Aβ production, including the proteolytic enzymes gamma- and β-secretases which generate Aβ from its precursor protein, APP (amyloid precursor protein). Aβ circulates in plasma, cerebrospinal fluid (CSF) and brain interstitial fluid (ISF) mainly as soluble Aβ40 Amyloid plaques contain both Aβ40 and Aβ42, while vascular amyloid is predominantly the shorter Aβ40. Several sequences of Aβ were found in both lesions. Generation of Aβ in the central nervous system may take place in the neuronal axonal membranes after APP-mediated axonal transport of β-secretase and presenilin-1.
Increases in either total Aβ levels or the relative concentration of both Aβ40 and Aβ42 (where the former is more concentrated in cerebrovascular plaques and the latter in neuritic plaques) have been implicated in the pathogenesis of both familial and sporadic Alzheimer's disease. Due to its more hydrophobic nature, the Aβ42 is the most amyloidogenic form of the peptide. However the central sequence KLVFFAE is known to form amyloid on its own, and probably forms the core of the fibril. One study further correlated Aβ42 levels in the brain not only with onset of Alzheimer's disease, but also reduced cerebrospinal fluid pressure, suggesting that a build-up or inability to clear Aβ42 fragments may play a role into the pathology.
The "amyloid hypothesis", that the plaques are responsible for the pathology of Alzheimer's disease, is accepted by the majority of researchers but is not conclusively established. An alternative hypothesis is that amyloid oligomers rather than plaques are responsible for the disease. Mice that are genetically engineered to express oligomers but not plaques (APPE693Q) develop the disease. Furthermore, mice that are in addition engineered to convert oligomers into plaques (APPE693Q X PS1ΔE9), are no more impaired than the oligomer only mice. Intra-cellular deposits of tau protein are also seen in the disease, and may also be implicated, as has aggregation of alpha synuclein.
Cancer
While Aβ has been implicated in cancer development, prompting studies on a variety of cancers to elucidate the nature of its possible effects, results are largely inconclusive. Aβ levels have been assessed in relation to a number of cancers, including esophageal, colorectal, lung, and hepatic, in response to observed reductions in risk for developing Alzheimer's disease in survivors of these cancers. All cancers were shown to be associated positively with increased Aβ levels, particularly hepatic cancers. This direction of association however has not yet been established. Studies focusing on human breast cancer cell lines have further demonstrated that these cancerous cells display an increased level of expression of amyloid precursor protein.
Down syndrome
Adults with Down syndrome had accumulation of amyloid in association with evidence of Alzheimer's disease, including declines in cognitive functioning, memory, fine motor movements, executive functioning, and visuospatial skills.
Formation
Aβ is formed after sequential cleavage of the amyloid precursor protein (APP), a transmembrane glycoprotein of undetermined function. APP can be cleaved by the proteolytic enzymes α-, β- and γ-secretase; Aβ protein is generated by successive action of the β and γ secretases. The γ secretase, which produces the C-terminal end of the Aβ peptide, cleaves within the transmembrane region of APP and can generate a number of isoforms of 30–51 amino acid residues in length. The most common isoforms are Aβ40 and Aβ42; the longer form is typically produced by cleavage that occurs in the endoplasmic reticulum, while the shorter form is produced by cleavage in the trans-Golgi network.
Genetics
Autosomal-dominant mutations in APP cause hereditary early-onset Alzheimer's disease (familial AD, fAD). This form of AD accounts for no more than 10% of all cases, and the vast majority of AD is not accompanied by such mutations. However, familial Alzheimer's disease is likely to result from altered proteolytic processing. This is evidenced by the fact that many mutations that lead to fAD occur near γ-secretase cleavage sites on APP. One of the most common mutations causing fAD, London Mutation, occurs at codon 717 of the APP gene, and results in a valine to isoleucine amino acid substitution. Histochemical analysis of the APP V717I mutation has revealed extensive Aβ pathology throughout neuroaxis as well as widespread cerebral amyloid angiopathy (CAA).
The gene for the amyloid precursor protein is located on chromosome 21, and accordingly people with Down syndrome have a very high incidence of Alzheimer's disease.
Structure and toxicity
Amyloid beta is commonly thought to be intrinsically unstructured, meaning that in solution it does not acquire a unique tertiary fold but rather populates a set of structures. As such, it cannot be crystallized and most structural knowledge on amyloid beta comes from NMR and molecular dynamics. Early NMR-derived models of a 26-aminoacid polypeptide from amyloid beta (Aβ 10–35) show a collapsed coil structure devoid of significant secondary structure content. However, the most recent (2012) NMR structure of (Aβ 1-40) has significant secondary and tertiary structure.Replica exchange molecular dynamics studies suggested that amyloid beta can indeed populate multiple discrete structural states; more recent studies identified a multiplicity of discrete conformational clusters by statistical analysis. By NMR-guided simulations, amyloid beta 1-40 and amyloid beta 1-42 also seem to feature highly different conformational states, with the C-terminus of amyloid beta 1-42 being more structured than that of the 1-40 fragment.
Low-temperature and low-salt conditions allowed to isolate pentameric disc-shaped oligomers devoid of beta structure. In contrast, soluble oligomers prepared in the presence of detergents seem to feature substantial beta sheet content with mixed parallel and antiparallel character, different from fibrils; computational studies suggest an antiparallel beta-turn-beta motif instead for membrane-embedded oligomers.
The suggested mechanisms by which amyloid beta may damage and cause neuronal death include the generation of reactive oxygen species during the process of its self-aggregation. When this occurs on the membrane of neurons in vitro, it causes lipid peroxidation and the generation of a toxic aldehyde called 4-hydroxynonenal which, in turn, impairs the function of ion-motive ATPases, glucose transporters and glutamate transporters. As a result, amyloid beta promotes depolarization of the synaptic membrane, excessive calcium influx and mitochondrial impairment. Aggregations of the amyloid-beta peptide disrupt membranes in vitro.
In AD brains, in addition to the characteristic aggregation of Aβ peptides that causes amyloid (senile) plaques, DNA damages also accumulate.Reactive oxygen species may be the major source of DNA damage in AD brains. CDK5 kinase phosphorylates APP at threonine 568 which stimulates Aβ peptide accumulation, and CDK5 activities also affect the DNA damage response, supporting the concept of a linkage of DNA damage with AD.
Intervention strategies
Researchers in Alzheimer's disease have identified several strategies as possible interventions against amyloid:
- β-Secretase inhibitors. These work to block the first cleavage of APP inside of the cell, at the endoplasmic reticulum.
- γ-Secretase inhibitors (e. g. semagacestat). These work to block the second cleavage of APP in the cell membrane and would then stop the subsequent formation of Aβ and its toxic fragments.
- Selective Aβ42 lowering agents (e. g. tarenflurbil). These modulate γ-secretase to reduce Aβ42 production in favor of other (shorter) Aβ versions.
β- and γ-secretase are responsible for the generation of Aβ from the release of the intracellular domain of APP, meaning that compounds that can partially inhibit the activity of either β- or γ-secretase are highly sought after. In order to initiate partial inhibition of β- and γ-secretase, a compound is needed that can block the large active site of aspartyl proteases while still being capable of bypassing the blood-brain barrier. To date, human testing has been avoided due to concern that it might interfere with signaling via Notch proteins and other cell surface receptors.
- Immunotherapy. This stimulates the host immune system to recognize and attack Aβ, or provide antibodies that either prevent plaque deposition or enhance clearance of plaques or Aβ oligomers. Oligomerization is a chemical process that converts individual molecules into a chain consisting of a finite number of molecules. Prevention of oligomerization of Aβ has been exemplified by active or passive Aβ immunization. In this process antibodies to Aβ are used to decrease cerebral plaque levels. This is accomplished by promoting microglial clearance and/or redistributing the peptide from the brain to systemic circulation. Antibodies that target Aβ and are currently in clinical trials included aducanumab, bapineuzumab, crenezumab, gantenerumab, lecanemab, and solanezumab. Amyloid beta vaccines that are currently in clinical trials include CAD106 and UB-311. However literature reviews have raised questions as to immunotherapy's overall efficacy. One such study assessing ten anti-Ab42 antibodies showed minimal cognitive protection and results within each trial, as symptoms were too far progressed by the time of application to be useful. Further development is still required for application to those who are presymptomatic to assess their effectiveness early into disease progression.
- Anti-aggregation agents such as apomorphine, or carbenoxolone. The latter has commonly been used as a treatment for peptic ulcers, but also displays neuroprotective properties, shown to improve cognitive functions such as verbal fluency and memory consolidation. By binding with high affinity to Aβ42 fragments, primarily via hydrogen bonding, carbenoxolone captures the peptides before they can aggregate together, rendering them inert, as well as destabilizes those aggregates already formed, helping to clear them. This is a common mechanism of action of anti-aggregation agents at large.
- Studies comparing synthetic to recombinant Aβ42 in assays measuring rate of fibrillation, fibril homogeneity, and cellular toxicity showed that recombinant Aβ42 had a faster fibrillation rate and greater toxicity than synthetic amyloid beta 1-42 peptide.
- Modulating cholesterol homeostasis has yielded results that show that chronic use of cholesterol-lowering drugs, such as the statins, is associated with a lower incidence of AD. In APP genetically modified mice, cholesterol-lowering drugs have been shown to reduce overall pathology. While the mechanism is poorly understood it appears that cholesterol-lowering drugs have a direct effect on APP processing.
- Memantine is an Alzheimer's disease drug which has received widespread approval. It is a non-competitive N-methyl-D-aspartate (NMDA) channel blocker. By binding to the NMDA receptor with a higher affinity than Mg2+ ions, memantine is able to inhibit the prolonged influx of Ca2+ ions, particularly from extrasynaptic receptors, which forms the basis of neuronal excitotoxicity. It is an option for the management of those with moderate to severe Alzheimer's disease (modest effect). The study showed that 20 mg/day improved cognition, functional ability and behavioural symptoms.
Measuring amyloid beta

Imaging compounds, notably Pittsburgh compound B, (6-OH-BTA-1, a thioflavin), can selectively bind to amyloid beta in vitro and in vivo. This technique, combined with PET imaging, is used to image areas of plaque deposits in those with Alzheimer's.
Post mortem or in tissue biopsies
Amyloid beta can be measured semiquantitatively with immunostaining, which also allows one to determine location. Amyloid beta may be primarily vascular, as in cerebral amyloid angiopathy, or in amyloid plaques in white matter.
One sensitive method is ELISA which is an immunosorbent assay which utilizes a pair of antibodies that recognize amyloid beta.
Atomic force microscopy, which can visualize nanoscale molecular surfaces, can be used to determine the aggregation state of amyloid beta in vitro.
Vibrational microspectroscopy is a label-free method that measures the vibration of molecules in tissue samples. Amyloid proteins like Aβ can be detected with this technique because of their high content of β-sheet structures. Recently, the formation of Aβ fibrils was resolved in different plaque-types in Alzheimer's disease, indicating that plaques transit different stages in their development.
Dual polarisation interferometry is an optical technique which can measure early stages of aggregation by measuring the molecular size and densities as the fibrils elongate. These aggregate processes can also be studied on lipid bilayer constructs.
See also
- TPM21
- Sylvain Lesné – Aβ*56