
| Chordoma | |
|---|---|
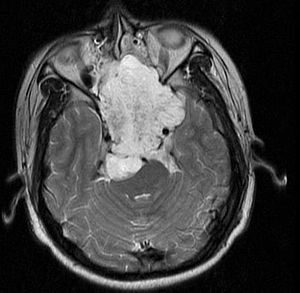 | |
| MRI of extensive clival chordoma in 17-year-old male patient, axial view. Tumor in the nasopharynx extending from nasal cavity to brainstem posteriorly is clearly visible. | |
| Specialty |
Oncology |
Chordoma is a rare slow-growing neoplasm thought to arise from cellular remnants of the notochord. The evidence for this is the location of the tumors (along the neuraxis), the similar immunohistochemical staining patterns, and the demonstration that notochordal cells are preferentially left behind in the clivus and sacrococcygeal regions when the remainder of the notochord regresses during fetal life.
In layman's terms, chordoma is a type of spinal cancer.
Presentation

Chordomas can arise from bone in the skull base and anywhere along the spine. The two most common locations are cranially at the clivus and in the sacrum at the bottom of the spine.
Sacral chordoma is presented with chronic low back pain.
Genetics
A small number of families have been reported in which multiple relatives have been affected by chordoma. In four of these families, duplication of the brachyury gene was found to be responsible for causing chordoma.
A possible association with tuberous sclerosis complex (TSC1 or TSC2) has been suggested.
Mechanism
- mTOR signaling is hyperactive in sporadic sacral chordomas: in one study 10 out of 10 sacral chordomas exhibited phosphorylation of Ribosomal protein s6 and EIF4EBP1 by immunohistochemistry
- Partial or complete PTEN (gene) deficiency is observed in nearly all sacral chordomas
- In a study of 49 chordomas Akt, TSC2, and EIF4EBP1 were phosphorylated in 92%, 96% and 98% of cases, respectively.
- In a tissue microarray containing 21 chordomas Platelet-derived growth factor receptor-beta (PDGFR-b), epidermal growth factor receptor (EGFR), KIT (CD117) and HER2 were detected in 100%, 67%, 33% and 0% of cases, respectively.
- The CDKN2A (p16) and CDKN2B (p15) loci on chromosome 9p21 are frequently deleted in chordomas Another study found CDKN2A immunoreactivity in just 4% of cases.
- 62% of chordomas express the High Molecular Weight Melanoma Associated Antigen, also known as Chondroitin sulfate proteoglycan 4 (CSPG4) which has been the target of immune therapy.
- In 2009, scientists discovered that an inherited gene duplication is responsible for the familial form of this disorder. Familial chordoma are rare, with an estimated rate of 0.4% in all Chordomas.
Diagnosis
In 2015 the first consensus guidelines for the diagnosis and treatment of chordoma were published in The Lancet Oncology. These tumors express brachyury and cytokeratin, which can be detected by immunohistochemistry.
Classification

There are three histological variants of chordoma: conventional, chondroid and dedifferentiated.
- The histological appearance of classical chordoma is of a lobulated tumor composed of groups of cells separated by fibrous septa. The cells have small round nuclei and abundant vacuolated cytoplasm, sometimes described as physaliferous (having bubbles or vacuoles).
- Chondroid chordomas histologically show features of both chordoma and chondrosarcoma.
Treatment
In most cases, complete surgical resection followed by radiation therapy offers the best chance of long-term control. Incomplete resection of the primary tumor makes controlling the disease more difficult and increases the odds of recurrence. The decision whether complete or incomplete surgery should be performed primarily depends on the anatomical location of the tumor and its proximity to vital parts of the central nervous system.
Chordomas are relatively radioresistant, requiring high doses of radiation to be controlled. The proximity of chordomas to vital neurological structures such as the brain stem and nerves limits the dose of radiation that can safely be delivered. Therefore, highly focused radiation such as proton therapy and carbon ion therapy are more effective than conventional x-ray radiation.
There are no drugs currently approved to treat chordoma, however a clinical trial conducted in Italy using a tyrosine kinase inhibitor imatinib demonstrated a modest response in some chordoma patients. The same group in Italy found that the combination of imatinib and sirolimus caused a response in several patients whose tumors progressed on imatinib alone. Erlotinib-like EGFR inhibitors have been also reported to be effective in chordoma. Although EGFR mutation is not present in chordoma, EGFR expression might predict response to erlotinib (as shown in report by Dr Sameer Rastogi).
A report of response to olaparib has been published.
Prognosis
In one study, the 10-year tumor free survival rate for sacral chordoma was 46%. Chondroid chordomas appear to have a more indolent clinical course.
Epidemiology
In the United States, the annual incidence of chordoma is approximately 1 in one million (300 new patients each year).
Sacral chordomas make up 2 to 4% of all primary bone tumours and 44% of all primary sacral tumours, thus making it the most common malignant sacral tumour. About 50 to 60% of chordomas are located in the sacrococcygeal region. Males aged between 40 and 50 years are twice as more common than women to get sacral chordoma.
There are currently no known environmental risk factors for chordoma. As noted above germline duplication of brachyury has been identified as a major susceptibility mechanism in several chordoma families.
While most people with chordoma have no other family members with the disease, rare occurrences of multiple cases within families have been documented. This suggests that some people may be genetically predisposed to develop chordoma. Because genetic or hereditary risk factors for chordoma may exist, scientists at the National Cancer Institute are conducting a Familial Chordoma Study to search for genes involved in the development of this tumor.
Society
Expert Recommendations for the Diagnosis and Treatment of Chordoma is a handbook produced by the Chordoma Foundation, which summarizes recommendations developed by a group of over 40 leading doctors who specialize in caring for chordoma patients. It is available electronically in English, Chinese, Italian, Dutch, and Spanish and hardcopies are available in English and Spanish.
Notable cases
NFL player Craig Heyward was treated for a chordoma in 1998, which ended his career. While initially thought to be successfully removed, the tumor returned in 2005, and caused Heyward's death in May 2006.
Pro skateboarder Ray Underhill, a member of the Powell-Peralta Bones Brigade, battled chordoma for two years before succumbing to his disease in August 2008.
Cary Tennis, the popular advice columnist for Salon, announced in his column of November 19, 2009, that he had been diagnosed with a chordoma.
Former Spanish footballer José Enrique was diagnosed with chordoma in May 2018 and underwent surgery to remove the tumour in June of that year. He announced in April 2019 that he had been given the all clear.
External links
- Images of Chordoma - mostly radiological (CT and MRI scans), one autopsy image
- Research information on chordoma (WikiGenes)