

β adrenergic receptor antagonists (also called beta-blockers or β-blockers) were initially developed in the 1960s, for the treatment of angina pectoris but are now also used for hypertension, congestive heart failure and certain arrhythmias. In the 1950s, dichloroisoproterenol (DCI) was discovered to be a β-antagonist that blocked the effects of sympathomimetic amines on bronchodilation, uterine relaxation and heart stimulation. Although DCI had no clinical utility, a change in the compound did provide a clinical candidate, pronethalol, which was introduced in 1962.
Development

History
The β-blockers are an immensely important class of drugs due to their high prevalence of use. The discovery of β-blockers reaches back to more than 100 years ago, when early investigators came up with the idea that catecholamines were binding selectively to receptor-like structures and that this was the cause of their pharmacological actions. In 1948, Raymond P. Ahlquist published a seminal paper concluding his findings, that there were two distinct receptors for catecholamine drugs, and they caused different responses in the heart muscle. He labeled them α-and β-adrenoceptors. These findings were soon to be a foundation for further research into drug development.
In the early 1960s, James Black, a Scottish pharmacologist, and associates of his at the Imperial Chemical Industries (ICI) in Great Britain were working on a series of β-adrenergic blocking compounds, pronethalol and propranolol. Dr. Black focused on developing a drug that would relieve the pain of angina pectoris, which results from oxygen deprivation in the heart. His plan was to create a drug that would decrease the heart's requirement for oxygen. He hypothesized that these compounds would lower the heart's oxygen consumption by interfering with the effects of catecholamines. In 1958, the pharmacological properties of dichloroisoproterenol (DCI) were described, a β-antagonist discovered a few years before by the Eli Lilly group. It was the synthesis of DCI which established that β-receptors could be chemically blocked and thus its existence could be confirmed. DCI had no clinical utility but a replacement of the 3,4-dichloro substituents, with a carbon bridge to form a naphtylethanolamine derivative, afforded a clinical candidate, pronethalol.
In April 1963, toxicity tests for pronethalol showed results of thymic tumours in mice. Nevertheless, it was launched under the trade name Alderlin, as the first clinically useful β-blocker. The launch took place in November 1963 when many small-scale clinical trials had proved their effectiveness in angina and certain types of arrhythmias. Pronethalol was only marketed for use in life-threatening situations. Dr. James Black went on to create another β-blocker, called propranolol; a non-selective β-blocker. Clinical trials started in the summer of 1964 and a year later, propranolol was launched under the trade name Inderal, only two and a half years after it had first been tested. It turned out to have a higher potency than pronethalol, with fewer side effects. Propranolol became the first major drug in the treatment of angina pectoris, since the introduction of coronary vasodilators, (such as nitroglycerin), almost 100 years earlier. Propranolol became a best-selling drug, used to treat a wide range of cardiovascular diseases such as arrhythmia, hypertension and hypertrophic cardiomyopathy.
The evolution of non-selective and selective β-blockers

By the time propranolol was launched, ICI was beginning to experience competition from other companies. This potential threat led to ongoing refinements in the pharmacologic structure of β-blockers and subsequent advances in drug delivery. ICI studied analogues further and in 1970 launched practolol (figure 4) under the trade name Eraldin. It was withdrawn from the market a few years later because of the severe side effects it caused, nevertheless it played a large role in the fundamental study of β-blockade and β-receptors. The withdrawal of Eraldin gave ICI the nudge to launch another β-blocker, atenolol, which was launched in 1976 under the trade name Tenormin. Atenolol is a selective β1-receptor antagonist and was developed for the purpose of obtaining the “ideal β-blocker”. It soon became one of the best-selling heart drug. ICI's β-blocker project was based on Ahlquist's dual receptor theory. The drugs that were the outcome of this project, from propranolol to atenolol, helped to establish the receptor theory among scientist and pharmaceutical companies.
The progress in β-blocker development led to the introduction of drugs with variety of properties. β-blockers were developed having a relative selectivity for cardiac β1-receptors (for example metoprolol and atenolol), partial adrenergic agonist activity (pindolol), concomitant α-adrenergic blocking activity (for example labetalol and carvedilol) and additional direct vasodilator activity (nebivolol). In addition, long-acting and ultra-short formulations of β-blockers were developed. In 1988, Sir James Black was awarded the Nobel Prize in Medicine for his work on drug development.






Mechanism of action
Pharmacokinetics
The β-adrenergic receptor antagonists all have similar therapeutic and pharmacodynamic actions in patients with cardiovascular disorders. They vary greatly in their pharmacokinetic properties, as they demonstrate a high range of values in plasma protein binding, the percent of drug eliminated by metabolism or unchanged in the urine and hepatic extraction ratio. Each of the β-blockers possesses at least one chiral centre and a high degree of enantioselectivity when binding to the β-adrenergic receptor. For those β-blockers containing a single chiral centre, the (-) enantiomer has a much higher affinity in binding to the β-adrenergic receptor than the (+) enantiomer. All β-blockers used systemically are delivered as racemate, except for timolol.
Binding to β adrenergic receptors

Three different types of β-adrenergic receptors have been identified by molecular pharmacology. β1-receptors are located in the heart and consist of about 75% of all β-receptors. β2-receptors can be found in the smooth muscles of vessels and the bronchies. β3-receptors are presumed to be involved in fatty acid metabolism and are found in the adipocytes. β-blockers cause a competitive inhibition of the β-receptor, which counters the effects of catecholamines.β1 and β2-receptors are G-protein coupled receptors, which couple to Gαs-proteins. When activated, it stimulates an increase in intracellular cAMP via the adenylyl cyclase. cAMP, which is the second messenger, then activates protein kinase A that phosphorylates the membrane's calcium channel and increases the entry of calcium into the cytosol. Protein kinase A also increases the release of calcium from the sarcoplasmic reticulum, which causes a positive inotropic effect. The phosphorylation of troponin I and phospholamban by protein kinase A causes the lusitropic effects of β-blockers. This increases the re-uptake of calcium by the sarcoplasmic reticulum.
β-blockers are sympatholytic drugs. Some β-blockers partially activate the receptor while preventing catecholamines from binding to the receptor, making them partial agonists. They provide a background of sympathetic activity, while preventing normal and enhanced sympathetic activity. These β-blockers possess intrinsic sympathomimetic activity (ISA). Some of them also possess what is called membrane-stabilizing activity (MSA) on myocardial muscle fibers.
Selectivity
β-blockers can be selective for either β1, β2 adrenergic receptor, or to be non-selective. By blocking β1 receptor it is possible to reduce heart rate, conduction of velocity and contractility. The blocking of β2 receptor promotes vascular smooth muscle contraction, which results in increase of peripheral resistance. Blockade of the β2 receptor effectively reduces the sympathetic activity, which results in reduce of the associated platelet-and coagulation activation. This is why a non-selective β-blocker treatment may result in a lower risk of both arterial and venous embolic events.
Synthesis


Synthesis for a standard β-blocker begins with the mono-alkylation of catechol to give an ether (see figure 4).
The fundamental step, and usually the last, in the synthesis of β-blockers consists of adding a propanolamine side chain. This can be done following two paths which both involve alkylation of an appropriate phenoxide with epichlorohydrin (ECH). The first way is shown as the upper way in figure 5. It consists of phenoxide reacting at the oxirane and resulting in an alkoxide, that displaces the adjacent chloride to form a new epoxide ring. The second way is shown as the lower route in figure 5. It consists of displacement of the halogen directly with a SN2 reaction to give the same glycidic ether. When following both pathways, the central chiral carbon preserves its configuration, which is an important part to consider when synthesizing enantiomerically defined drugs. The ring opening of the epoxide ring in glycidic ether is done with an appropriate amine, such as isopropyl amine or tert-butylamine, and leads to the aryloxypropanolamine compound that consist of a secondary amine. This amine is typically known as the structural requirement for the β-adrenergic blocking activity.
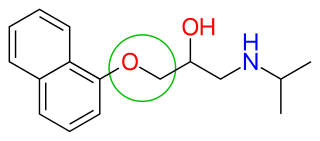
(S)-propranolol

Propranolol exist in two different enantiomers, (S)-(−)- and (R)-(+)-enantiomers. The (S)-isomer is 100-fold more potent than the (R)-isomer and that is the general rule for most β-blockers. It is possible to produce the (S)-propranolol enantiomer from α-naphthol and 3-bromopropanol as seen in figure 6. α-Naphthol and 3-bromopropanol are refluxed for 6 hours to give alcohol. The alcohol is oxidized by using 2-Iodoxybenzoic acid (IBX) to give aldehyde. The aldehyde is subjected to L-proline catalyzed asymmetric α-aminoxylation and a reduction is made with NaBH4 in methanol. A diol is obtained by Pd/C-catalyzed hydrogenolysis. Finally the diol is converted to epoxide using the Mitsunobu reaction and stirred with isopropyl amine in CH2Cl2 to give (S)-propranolol.
Structure-activity relationship (SAR)
β-blockers' binding site to the receptor is the same as for endogenous catecholamines, such as noradrenaline and adrenaline. This binding is based on hydrogen bonds between the β-blocker and the receptor, and therefore not based on covalent bonds, which results in the reversibility of the binding. A significant step in the development of β adrenergic antagonists was the discovery that an oxymethylene bridge (—OCH2—, figure 7) could be inserted into the arylethanolamine structure of pronethalol to produce propranolol. Propranolol is an aryloxypropanolamine, which are more potent β-blockers than arylethanolamines. Today, most of the β-blockers used clinically are aryloxypropanolamines. The length of the side chain is increased when an oxymethylene bridge is introduced. It has been shown that the side chains of aryloxypropanolamine can adopt a conformation that puts the hydroxyl and amine groups in more or less the same position as with beta blocker that do not have this group as a part of the side chain.
After the release of propranolol, relative lipophilicity of β-blockers as a significant factor in their varied and complex pharmacology, became an important factor. It was suspected that propranolol's centrally induced side effects could be due to its high lipophilicity. Thus, it was focused on synthesizing analogues with hydrophilic moieties, favourably placed to see if the side effects would decrease. Selecting para-acylamino groups as the hydrophilic moiety, scientists synthesized a group of para-acylphenoxyethanol and propanolamines, and selected practolol for clinical trials. Practolol had one property not previously seen with β-blockers, it exhibited cardioselectivity (β1 selectivity). Studies from practolol showed that moving the acylamino group to meta or ortho positions, on the benzene ring, caused a loss of selectivity but not loss of the β-blockade itself. This illustrated the significance of para-substitution for β1 selectivity of β-blockers.

Figure 8 shows the structure-activity relationship (SAR) for β-blockers. For the function of a β-blocker it's essential for the compound to contain an aromatic ring and a β-ethanolamine. The aromatic ring can either be benzoheterocyclic (such as indole) or heterocyclic (such as thiadiazole). This is mandatory. The side chains can be variable:
- The X part of the side chain can either be directly linked to the aromatic ring or linked through a —OCH2— group
- When X is —CH2CH2—, —CH=CH—, —SCH2— or —NCH2—, there is little or no activity
- The R1 group can only be a secondary substitution and branched is the optimal choice
- Alkyl (—CH3) substituents on the α, β or γ carbon (if X = —OCH2—) lower beta blockade, especially at the α carbon
The general rule for aromatic substitution is: ortho > meta > para. This gives non-selective β-blockers. Large para-substituents usually decrease activity but large ortho-groups retain some activity. Polysubstitution on carbon 2 and 6 makes the compound inactive but when the substitution is on carbon 3 and 5 there's some activity. For the highest cardioselectivity, the substituents should be as following: para > meta > ortho. All the β-blockade is in one isomer, (S)-aryloxypropylamine and (R)-ethanolamine.
Clinical use
Cardiovascular indications
For decades β-blockers have been used in cardiovascular medicine. They have proved to reduce morbidity and mortality. In acute coronary syndrome, β-blockers have been recommended as a class I-A indication in clinical practice guidelines, because the treatment decreases the mortality rate. β-blockers, along with calcium channel blockers, reduce the workload of the heart and its oxygen requirement. β-blockers are sometimes used in a combination therapy to treat angina, if a β-blocker doesn't work well enough on its own. They are used as anti-arrhythmic drugs in patients with hyperthyroidism, cardiac dysrhythmia, atrial fibrillation, atrial flutter and ventricular tachycardia. The treatment with β-blockers reduces the incidence of sudden heart failure when the patient has already had a myocardial infarction. The reason is probably because of their anti-arrhythmic effects and also anti-ischemic effects. A β-blocker therapy is also useful in myocardial infarction, independent to heart failure. The therapy has been very helpful for high-risk patients. Although beta-blockers effectively lower blood pressure, they are not recommended as a first-line agent in the treatment of hypertension, as thiazides diuretics, ACE inhibitors, and calcium channel blockers show greater benefit. Therefore, β-blockers are usually used alongside other blood pressure medications such as calcium channel blockers. They also have an effect on cardiomyopathy, postural orthostatic tachycardia syndrome and portal hypertension, to name a few.
Other indications
There are few diseases, other than cardiovascular diseases, that β-blockers have a clinical effect on. These diseases are mentioned in the following sub-chapters. In addition, there are diseases which β-blockers have a clinical effect but are not the first choice of treatment. They won't be mentioned in the sub-chapters.
Essential tremor
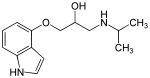
When symptoms of the essential tremors are considerably high, non-selective β-blockers are an important treatment option and usually the first choice. Studies have shown that propranolol did reduce symptoms the most in that category. The β-blockers can be used alone or in a combination.
Glaucoma
Glaucoma is caused by high intra-ocular pressure (IOP). β-blockers reduce IOP and are the most common therapy. Most of the patients, who use the topical β-blockers, need adjunctive therapy to achieve a target IOP lowering. One of the most used drug in adjunctive therapy is dorzolamide.
Teratogenicity
Hypertension is reported to complicate one out of ten pregnancies, which makes it the most common medical disorder in pregnancy. It is important to have a correct diagnosis of hypertension during pregnancy, with the emphasis on differentiating pre-existing hypertension from pregnancy induced hypertension (gestational and the syndrome of pre-eclampsia). During pregnancy, the challenge is to determine when to use antihypertensive medications and which level of blood pressure to target. A balance has to be found between the potential risk to the health of the baby related to drug-exposure and the risk to the mother and baby due to an untreated medical condition (severe hypertension).
Antihypertensive drug use during pregnancy is relatively common and increasing. Only a small proportion of available antihypertensive drugs have been tested in pregnant women, and many are contraindicated. It is important to make the exposure of medications to the baby as small as possible. It is not clear if treating women who have mild or moderate hypertension during pregnancy with anti-hypertensive medication is beneficial.
The most common first trimester antihypertensive are β-blockers. The consequences of treatment with β-blocker during pregnancy are disputable. Some studies report a connection between β-blocker treatment and small for-gestational-age (SGA) of newborns and pre-term birth, while others do not. Based on meta-analyses, first trimester oral β-blocker use showed no increase in odds of major congenital anomalies. However, analyses examining organ-specific malformations observed increased odds of cardiovascular defects, cleft lip and neural tube defects. The U.S. Food and Drug Administration (FDA) categorises β-blockers into different pregnancy categories depending on the safety of the drugs and they range from category B to D, that is, no β-blockers is completely safe for using during pregnancy
| Non-selective | FDA category | β1-selective | FDA category |
|---|---|---|---|
| Labetalol | C | Atenolol | D |
| Pindolol | B | Acebutolol | B |
| Propranolol | C | Bisoprolol | C |
| Timolol | C | Nebivolol | C |
| Sotalol | B | Metoprolol | C |