
Four drugs from the class of direct Xa inhibitors are marketed worldwide. Rivaroxaban (Xarelto) was the first approved FXa inhibitor to become commercially available in Europe and Canada in 2008. The second one was apixaban (Eliquis), approved in Europe in 2011 and in the United States in 2012. The third one edoxaban (Lixiana, Savaysa) was approved in Japan in 2011 and in Europe and the US in 2015.Betrixaban (Bevyxxa) was approved in the US in 2017.
History
Heparin
Heparin was discovered by Jay McLean and William Henry Howell in 1916, it was first isolated from a canine liver, which in Greek translates to hepar. Heparin targets multiple factors in the blood coagulation cascade, one of them being FXa. At first, it had many side effects but for the next twenty years, investigators worked on heparin to make it better and safer. It entered clinical trials in 1935 and the first drug was launched in 1936. Chains of natural heparin can vary from 5.000 to 40.000 Daltons. In the 1980s Low molecular weight heparin (LMWH) were developed and they only contain chains with an average molecular weight of less than 8.000 Da.
Warfarin

In the 1920s there was an outbreak of a mysterious haemorrhagic cattle disease in Canada and the northern United States. The disease was named sweet clover disease because the cattle had grazed on sweet clover hay. It wasn't until ten years after the outbreak, that a local investigator, Karl P. Link and his student Wilhelm Schoeffel started an intense investigation to find the substance causing the internal bleeding. It took them 6 years to discover dicoumarol, the causing agent. They patented the right for the substance and in 1945 Link started selling a coumarin derivative as a rodenticide. He and his colleagues worked on several variations and ended up with a substance they named warfarin in 1948. It wasn't until 1954 that it was approved for medicinal use in humans making warfarin the first oral anticoagulant drug.
Need for newer and better oral drugs
Warfarin treatment requires blood monitoring and dose adjustments regularly due to its narrow therapeutic window. If supervision isn't adequate warfarin poses a threat in causing, all too frequent, haemorrhagic events and multiple interactions with food and other drugs. Currently, the main problem with low molecular weight heparin (LMWH) is the administration route, as it has to be given subcutaneously. Because of these disadvantages there has been an urgent need for better anticoagulant drugs. For a modern society, convenient and fast drug administration is the key to a good drug compliance. In 2008 the first direct Xa inhibitor was approved for clinical use. Direct Xa inhibitors are just as efficacious as LMWH and warfarin but they are given orally and don't need as strict monitoring. Other Xa inhibitors advantages are rapid onset/offset, few drug interactions and predictable pharmacokinetics. The rapid onset/offset effect greatly reduces the need for “bridging” with parenteral anticoagulants after surgeries. Today there are four factor Xa inhibitors marketed: rivaroxaban, apixaban, edoxaban and betrixaban.
Antistasin and tick anticoagulant peptide (TAP)
Factor Xa was identified as a promising target for the development of new anticoagulants in the early 1980s. In 1987 the first factor Xa inhibitor, the naturally occurring compound antistasin, was isolated from the salivary glands of the Mexican leech Haementeria officinalis. Antistasin is a polypeptide and a potent Xa inhibitor. In 1990 another naturally occurring Xa inhibitor was isolated, tick anticoagulant peptide (TAP) from extracts of the tick Ornithodoros moubata. TAP and antistasin were used to estimate factor Xa as a drug target.
Mechanism of action

Blood coagulation is a complex process by which the blood forms clots. It is an essential part of hemostasis and works by stopping blood loss from damaged blood vessels. At the site of injury, where there is an exposure of blood under the endothelium, the platelets gather and immediately form a plug. That process is called primary hemostasis. Simultaneously, a secondary hemostasis occurs. It is defined as the formation of insoluble fibrin by activated coagulation factors, specifically thrombin. These factors activate each other in a blood coagulation cascade that occurs through two separate pathways that interact, the intrinsic and extrinsic pathway. After activating various proenzymes, thrombin is formed in the last steps of the cascade, it then converts fibrinogen to fibrin which leads to clot formation. Factor Xa is an activated serine protease that occupies a key role in the blood coagulation pathway by converting prothrombin to thrombin. Inhibition of factor Xa leads to antithrombotic effects by decreasing the amount of thrombin. Directly targeting factor Xa is suggested to be an effective approach to anticoagulation.
Development

In 1987 antistasin was tested as the first direct Xa inhibitor. Antistasin is a protein made up of 119 amino acid residues, of which 20 are cysteines involved in 10 disulfide bonds. It acts as a slow, tight-binding inhibitor of factor Xa with a Ki value of 0.3–0.6 nM but it also inhibits trypsin. Recombinant Antistasin can be produced by genetically modified yeast, saccharomyces cerevisiae. Another natural occurring direct Xa-inhibitor, the tick anticoagulant peptide (TAP), was discovered in 1990. It is a single-chain, 60 amino acid peptide and like antistasin it is a slow, tight-binding inhibitor with a similar Ki value (~0.6 nM).
These two proteins were mostly used to validate factor Xa as a drug target. Animal studies suggested direct Xa-inhibition to be a more efficient approach to anticoagulation compared to direct thrombin inhibitors, especially offering a wider therapeutic window and reducing the risk of rebound thrombosis, (increase in thromboembolic events occurring shortly after the withdrawal of an antithrombotic medication) compared to direct and indirect thrombin inhibitors.
During the 1990s several low-molecular-weight substances were developed, such as DX-9065a and YM-60828.

DX-9065a was the first synthetic compound that inhibited FXa without inhibiting thrombin. That was attained by inserting a carboxyl group which seemed to be the most important moiety for a selective binding to FXa. Those early developed small molecules yet had amidine-groups or even higher-basic functions, which were thought to be necessary as mimics for an arginine residue in prothrombin, the natural substrate of factor Xa. Nevertheless, these basic functions are also related to a very poor oral bioavailability (e.g. 2–3% for DX-9065a).
In 1998 Bayer Healthcare, a pharmaceutical company started searching for low-molecular-weight direct factor Xa inhibitors with higher oral bioavailability. High-throughput screening and further optimisation at first lead to several substances from the class of isoindolinones demonstrating that much less basic substances can also act as potent Xa inhibitors to an IC50 value of up to 2 nM. Although isoindolinones have a better oral bioavailability than the original compounds it wasn't sufficient enough. However, the project later lead to the class of n-aryloxazolidinones that provides substances with both high potency of inhibiting factor Xa and high bioavailability. One compound of this class, Rivaroxaban (IC50 = 0.7 nM, bioavailability: 60%), was granted marketing authorization for the prevention of venous thromboembolism in Europe and Canada in September 2008.
Chemistry
Factor Xa: Structure and binding sites

Factors IIa, Xa, VIIa, IXa and XIa are all proteolytic enzymes that have a specific role in the coagulation cascade. Factor Xa (FXa) is the most promising one due to its position at the intersection of the intrinsic and extrinsic pathway as well as generating around 1000 thrombin molecules for each Xa molecule which results in a potent anticoagulant effect. FXa is generated from FX by cleavage of a 52 amino acid activation peptide, as the "a" in factor Xa means activated. FXa consists of 254 amino acid catalytic domain and is also linked to a 142 amino acid light chain. The chain contains both GLA domain and two epidermal growth factor domains (EGF like domains).
The active site of FXa is structured to catalyze the cleavage of physiological substrates and cleaves PhePheAsnProArg-ThrPhe and TyrIleAspGlyArg-IleVal in prothrombin. FXa has four so-called pockets which are targets for substrates to bind to factor Xa. These pockets are lined up by different amino acids and Xa inhibitors target these pocket when binding to factor Xa. The two most relevant pockets regarding affinity and selectivity for the Xa inhibitors are S1 and S4.
S1: The S1 pocket is a hydrophobic pocket and contains an aspartic acid residue (Asp-189) which can serve as a recognition site for a basic group. FXa has a residual space in the S1 pocket and is lined by residues Tyr-228, Asp-189 and Ser-195.
S2: The S2 pocket is a small and shallow pocket. It merges with the S4 pocket and has room for small amino acids. Tyr-99 seems to block access to this pocket, so this pocket is not as important as S1 and S4.
S3: The S3 pocket is located on the rim of the S1 pocket and is flat and exposed to the solvent. This pocket is not as important as S1 and S4.
S4: The S4 pocket is hydrophobic in nature and the floor of the pocket is formed by Trp-215 residue. The residues Phe-174 and Tyr-99 of FXa join Trp-215 to form an aromatic box that is able to bind aliphatic, aromatic and positively charged fragments. Because of the binding to positively charged entities, it can be described as a cation hole.
Chemical structure and properties of direct Xa inhibitors
| Rivaroxaban | Apixaban | Edoxaban | |
|---|---|---|---|
| MW (g/mol) | 436 | 460 | 548 |
| Molecular formula | C19H18ClN3O5S | C25H25N5O4 | C24H30ClN7O4S |
| Shape | L | L | L |
| Ki | 0.4 nM | 0,08 nM | 0.561 nM |
| IC50 | 0.7 nM | N/A | N/A |
| Oral bioavailability (%) | 66–100 (dose-dependent) | 50 | 62 |

Binding of Xa inhibitors to factor Xa
The Xa inhibitors all bind in a so-called L-shape fashion within the active site of factor Xa. The key constituents of the factor Xa are the S1 and S4 binding sites. It was first noted that the natural compounds, antistasin and TAP, which possess highly polar and therefore charged components bind to the target with some specificity. That's why newer drugs were designed with positively charged groups but those resulted in poor bioavailability. Nowadays marketed Xa inhibitors, therefore contain an aromatic ring with various moieties attached for different interactions with the S1 and S4 binding sites. This also ensures good bioavailability as well as maintaining firm binding strength. The Xa inhibitors currently on market today, therefore rely on hydrophobic and hydrogen bonding instead of highly polar interactions.
Antistasin binding to factor Xa
Antistasin contains an N- and a C-terminal domain which are similar in their amino acid sequences with ~40% identity and ~56% homology. Each of them contains a short β-sheet structure and 5 disulfide bonds. Only the N-terminal domain is necessary to inhibit Xa while the C-terminal domain does not contribute to the inhibitory properties due to differences in the 3 dimensional structure, even though the C-terminal domain has a strongly analogue pattern to the actual active site.
The interaction of antistasin with FXa involves both the active site and the inactive surface of FXa. The reactive site of antistasin formed by Arg-34 and Val-35 in the N-terminal domain suits the binding site of FXa, most likely the S1 pocket. At the same time, Glu-15 located outside the reactive site of antistasin fits to positively charged residues on the surface of FXa. The multiple binding is thermodynamically advantageous and leads to sub-nanomolar inhibition (Ki = 0.3–0.6 nM).
DX-9065a binding to factor Xa
DX-9065a, the first small molecule direct Xa-inhibitor, is an amidinoaryl derivate with a molecular weight of 571.07g/mol. Its positively charged amidinonaphtalene group forms a salt bridge to the Asp-189 residue in the S1 pocket of FXa. The pyrrolidine ring fits between Tyr-99, Phe-174 and Trp-215 in the S4 pocket of FXa.
Unlike older drugs, e.g. heparin, DX-9065a is selective for FXa compared to thrombin even though FXa and thrombin are similar in their structure. This is caused by a difference in the amino acid residue in the homologue position 192. While FXa has a glutamine residue in that position, thrombin has a glutamic acid that causes electrostatic repulsion with the carboxyl group of DX-9065a. In addition, a salt bridge between Glu-97 of thrombin and the amidine group fixed in the pyrrolidine ring of DX-9065a reduces the flexibility of the DX-9065a molecule, which now cannot rotate enough to avoid the electrostatic clash. That's why the IC50 value for thrombin is >1000µM while the IC50 value for FXa is 0,16µM.

Rivaroxaban binding to factor Xa
Rivaroxaban binding to FXa is mediated through two hydrogen bonds to the amino acid Gly-219. These two hydrogen bonds serve an important role directing the drug into the S1 and S4 subsites of FXa. The first hydrogen bond is a strong interaction which comes from the carbonyl oxygen of the oxazolidinone core of rivaroxaban. The second hydrogen bond is a weaker interaction and comes from the amino group of the clorothiophene carboxamide moiety.
These two hydrogen bonds result in the drug forming an L-shape and fits in the S1 and S4 pockets. The amino acids residues Phe-174, Tyr-99, and Trp-215 form a narrow hydrophobic channel that is the S4 binding pocket. The morpholinone part of rivaroxaban is “sandwiched” between amino acids Tyr-99 and Phe-174 and the aryl ring of rivaroxaban is oriented perpendicularly across Trp-215. The morpholinone carbonyl group does not have a direct interaction to the FXa backbone, instead, it contributes to a planarization of the morpholinone ring and therefore supports rivaroxaban to be sandwiched between the two amino acids.
The interaction between the chlorine substituent of the thiophene moiety and the aromatic ring of Tyr-228, which is located at the bottom of the S1, it is very important due to the fact that it obviates the need for strongly basic groups for high affinity for FXa. This enables rivaroxaban, which is non-basic, to achieve good oral bioavailability and potency.

Apixaban binding to factor Xa
Apixaban shows a similar binding mode as rivaroxaban and forms a tight inhibitor-enzyme complex when connected to FXa. The p-methoxy group of apixaban connects to S1 pocket of FXa but does not appear to have any interaction with any residues in this region of FXa. The pyrazole N-2 nitrogen atom of apixaban interacts with Gln-192 and the carbonyl oxygen interacts with Gly-216. The phenyl lactam group of apixaban is positioned between Tyr-99 and Phe-174 and due to its orientation, it is able to interact with Trp-215 of the S4 pocket. The carbonyl oxygen group of the lactam moiety interacts with a water molecule and does not seem to interact with any residues in the S4 pocket.

Structure-activity-relationship (SAR)
An important part of designing a compound, that is an ideal inhibitor to a certain target, is to understand the amino acid sequence of the target site for the compound to bind to. Modelling both prothrombin and FXa makes it possible to deduct the difference and identify the amino acids at each binding site. At the bottom of the S1 pocket on FXa the binding amino acid is Asp-189 which amidine moieties can bind to. After X-raying the binding site of FXa, it was revealed that the S1 pocket had a planar shape, meaning that a flat amidinoaryl group should bind to it without steric hindrance.
Modern direct Xa inhibitors are L-shaped molecules whose ends fit perfectly in the S1 and S4 pockets. The long side of the L-shape has to conform to a highly-specific tunnel within the targets active site. To accomplish that, this part of the molecules is designed to have little formal interactions with FXa in that region. As there is no specific bonding, the fit of these agents between the pockets of FXa increases the total specificity of the drugs to the FXa molecule. The interaction between the S1 pocket of FXa and the inhibitor can be both ionic or non-ionic, which is important because it allows the design of the moiety to be adjusted to increase oral bioavailability. Previously designed compounds were charged molecules that are not absorbed well in the gastrointestinal tract and therefore did not reach high serum concentrations. The newer drugs have a better bioavailability as they are not charged and have a non-ionic interaction to the S1 pocket.
Rivaroxaban
During the SAR development of rivaroxaban, researchers realized that adding a 5-chlorothiophene-2-carboxamide group to the oxazolidonine core could increase the potency by 200 fold, which had previously been too weak for medical use. In addition to this discovery, a clear preference for the (S)-configuration was confirmed. This compound had a promising pharmacokinetical profile and did not contain a highly basic amidine group, but that had previously been considered important for the interaction with the S1 pocket. These findings lead to extensive SAR (structure-activity relationship) researches. During the SAR testing, R1 was defined as the most important group for potency. Pyrrolidinone was the first R1 functional group to significantly increase the potency but further researches revealed even higher potency with a morpholinone group instead. Groups R2 and R3 had hydrogen or fluorine attached and it was quickly assessed that having hydrogen resulted in highest potency. Groups R2 and R3 were then substituted for various groups, which were all less potent than the hydrogen, so hydrogen was the final result. As the chlorothiophene moiety had an inadequate water solubility, substituting it with another group was attempted but was unsuccessful. The chlorothiophene moiety binds to Tyr-228 at the bottom of the S1 pocket, making it a key factor regarding binding to FXa. Rivaroxaban has both high affinity and good bioavailability.

Apixaban

During the SAR development of apixaban there were three groups that needed to be tested to attain maximum potency and bioavailability. The first group to be tested was the non-active site as it needs to be stabilized before SAR testing on the p-methoxyphenyl group (S1 binding moiety). There are several of groups that increase the potency of the compound, mostly amides, amines and tetrazoles but also methylsulfonyl and trifluoromethyl groups. Of these groups, carboxamide has the greatest binding and had similar clotting activity as the compounds.
In dog testing, this compound with a carboxamide group called 13F, showed a great pharmacokinetical profile, a low clearance and adequate half-life and volume of distribution. Due to the success of finding a stabilizing group, SAR research for S1 binding moiety (p-methoxyphenyl) was discontinued. In the S4 binding group, N-methylacetyl and lactam analogues proved to have a very high binding affinity for FXa, showed great clotting and selectivity versus other proteases. Orientation turned out to be important as N-methyl acetyl, compared to acetamide, had a 300 fold lower binding ability to FXa due to unfavorable planarity close to the S4 region binding site.
Synthesis
Rivaroxaban
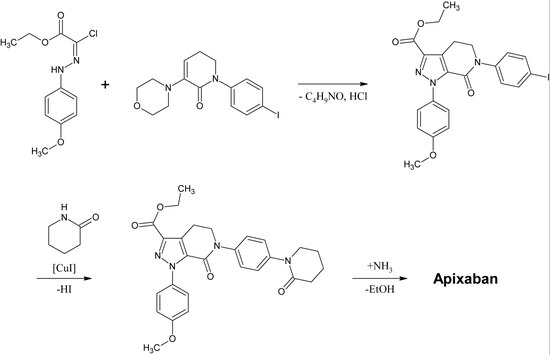
Rivaroxaban chemically belongs to the group of n-aryloxazolidinones. Other drugs of that group are linezolid and tedizolid, both of whom are antibiotics. A synthesis of n-aryloxazolidinones starting with an O-silyl protected ethyl(2,3-dihydroxypropyl)-carbamate was published in 2016. In a one-pot reaction the carbamate cyclisizes to a 2-oxazolidone ring under slightly basic conditions while simultaneously the oxazolidone nitrogen is arylized by copper-catalization. For rivaroxaban in particular, 3-morpholinone substitutes the iodine in p-position of the benzene ring by copper-catalization. Afterwards, the silyl protecting group is removed and the resulting alcohol is replaced by an amino group which is then acylated in the last step.
An industrial preparation of rivaroxaban was registered as a patent by Bayer Healthcare in 2005. It starts from N-(4-aminophenol)-morpholinone which is alkylated by a propylene oxide derivate that also contains a primary amine involved in a phthalimide protection group. Next, a phosgene equivalent is added to form the 2-oxazolidone ring and the phthalimide is removed. The free amine can now be acylated which leads to rivaroxaban.
However, according to the patent the synthesis has “various disadvantages in the reaction management which has particularly unfavourable effects for preparation“. The patent also explains another synthesis starting from a chlorothiophene derivate that would be more suitable for the industrial process but points out that toxic solvents or reagents have to be removed from the final product. Therefore, this way is not an alternative.
Various other synthesis pathways of rivaroxaban have been described.
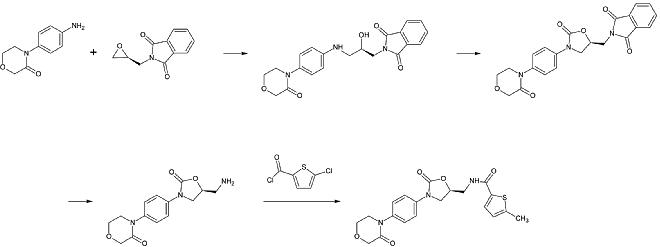
Apixaban
The first full synthesis of apixaban was published in 2007. The key step of this reaction is a (3+2)cycloaddition of a p-methoxyphenylchlorohydrazon derivate and a p-iodophenyl-morpholin-dihydropyridin derivate. After the following elimination of HCl and morpholine, the iodine is substituted by 2-piperidinone by copper-catalization and the ethyl esther is converted to an amide (aminolysis). This reaction was registered as a patent in 2009.
Clinical use
Direct factor Xa inhibitors are being used clinically and their usage is constantly increasing. They are gradually taking over warfarin usage and low molecular weight heparins (LMWH). Indication for Xa inhibitors is preventing deep vein thrombosis (DVT) which can lead to pulmonary embolism. It is also used to treat atrial fibrillation to lower the risk of stroke caused by a blood clot. Another indication is a prophylactic treatment for blood clotting (thrombosis) due to atherosclerosis. Rivaroxaban was the first FXa inhibitor on the market and then followed by apixaban, edoxaban and betrixaban.
| Rivaroxaban | Apixaban | Edoxaban | Betrixaban | |
|---|---|---|---|---|
| Brand name | Xarelto | Eliquis | Savaysa, Lixiana | Bevyxxa |
| Developer and producer | Bayer | Pfizer | Daiichi Sankyo | Portola Pharmaceuticals |
Pharmacokinetics
| Rivaroxaban | Apixaban | Edoxaban | |
|---|---|---|---|
| Metabolism | CYP3A4/5 (major), CYP2J2 (minor) | CYP3A4 (major), CYP1A2, 2C8, 2C19, 2J2 (all minor) | CYP34A (major) |
| Protein binding (%) | 92–95 | 87 | 55 |
| Half life (hrs) | 5–9 | 6–12 | 5–11 |
| Elimination | Renal (66%; 36% as unchanged drug) | Renal (27%), fecal | Renal (35%) |
| Absorption (Tmax) | 2–4 hours | 3–4 hours | 1–2 hours |
| Distribution (L) | 50 | 21–61 | 107 |
| Renal clearance (L/hr) | 2.4 | 7.5 | 11 |
Future perspectives
Direct Xa inhibitors in clinical trials
Rivaroxaban, apixaban, edoxaban and betrixaban are already on the market. As of October 2016, several new direct Xa inhibitors have entered clinical trials. These are letaxaban from Takeda and eribaxaban from Pfizer.
Antidotes
Andexxa (Andexanet alfa) from Portola Pharmaceuticals is a recombinant protein that is given intravenously. It works as an antidote to all direct and indirect FXa inhibitors. Andexxa acts as a decoy receptor for Xa inhibitors.