
| Familial adenomatous polyposis | |
|---|---|
| Other names | FAP |
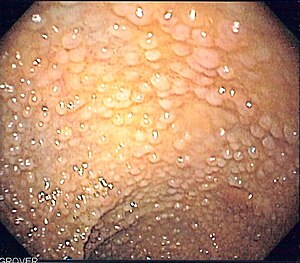 | |
| Endoscopic image of sigmoid colon of patient with familial adenomatous polyposis | |
| Specialty | Gastroenterology, Oncology |
| Complications | Colorectal cancer |
| Usual onset | <35 years of age |
| Duration | Lifelong |
| Types | Classical or attenuated |
| Causes | APC gene mutation |
| Diagnostic method |
Colonoscopy Genetic testing |
| Differential diagnosis | Lynch syndrome, MUTYH-associated polyposis |
| Treatment | Colonoscopy Polypectomy Upper endoscopy Colectomy |
| Frequency | 1 in 10,000 - 15,000 |
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited condition in which numerous adenomatous polyps form mainly in the epithelium of the large intestine. While these polyps start out benign, malignant transformation into colon cancer occurs when they are left untreated. Three variants are known to exist, FAP and attenuated FAP (originally called hereditary flat adenoma syndrome) are caused by APC gene defects on chromosome 5 while autosomal recessive FAP (or MUTYH-associated polyposis) is caused by defects in the MUTYH gene on chromosome 1. Of the three, FAP itself is the most severe and most common; although for all three, the resulting colonic polyps and cancers are initially confined to the colon wall. Detection and removal before metastasis outside the colon can greatly reduce and in many cases eliminate the spread of cancer.
The root cause of FAP is understood to be a genetic mutation—a change in the body's tumour suppressor genes that prevent development of tumours. The change allows numerous cells of the intestinal wall to develop into potentially cancerous polyps when they would usually reach the end of their life; inevitably one or more will eventually progress and give rise to cancer (7% risk by age 21, rising to 87% by age 45 and 93% by age 50). These gene changes do not trigger cancer, but rather, they reduce the body's ability to prevent cells from becoming cancerous. Even with the gene change, it may still take time before a cell actually does develop that is cancerous as a result, and the gene may in some cases still partially operate to control tumours, therefore cancer from FAP takes many years to develop and is almost always an adult-onset disease.
The second form of FAP, known as attenuated familial adenomatous polyposis has the APC gene functional but slightly impaired. It is therefore somewhat able to operate as usual. Attenuated FAP still presents a high 70% lifetime risk of cancer (as estimated), but typically presents with far fewer polyps (typically 30) rather than the hundreds or thousands usually found in FAP, and arises at an age when FAP is usually no longer considered likely—typically between 40 and 70 years old (average 55) rather than the more usual 30s upward. Because it has far fewer polyps, options for management may be different.
The third variant, autosomal recessive familial adenomatous polyposis or MUTYH-associated polyposis, is also milder and, as its name suggests, requires both parents to be 'carriers' to manifest the condition.
In some cases FAP can manifest higher in the colon than usual (for example, the ascending colon, or proximal to the splenic flexure, or in the stomach or duodenum) where they show no symptoms until cancer is present and greatly advanced. APC mutations have been linked to certain other cancers such as thyroid cancer. As the mutation causing FAP is autosomal dominant, it can be inherited directly from either parent to a child. A genetic blood test of the APC gene exists that can determine whether it is present, and therefore can predict the possibility of FAP. Individuals at risk (due to family links or genetic testing) are usually offered routine monitoring of the intestinal tract every 1–3 years for life, from puberty for FAP and early adulthood for attenuated forms. Colon resection surgery is recommended if numerous colon polyps are found due to high risk of early death from colon cancer. International polyposis registries exist that track known cases of FAP or APC gene defects, for research and clinical purposes. Mutation of APC also occurs commonly in incident cases of colorectal carcinoma, emphasizing its importance in this form of cancer.
Signs and symptoms
From early adolescence, patients with this condition gradually (and much of the time asymptomatically) develop hundreds to thousands of colorectal polyps (and sometimes polyps elsewhere)—small abnormalities at the surface of the intestinal tract, especially in the large intestine including the colon or rectum. These may bleed, leading to blood in the stool. If the blood is not visible, it is still possible for the patient to develop anemia due to gradually developing iron deficiency. If malignancy develops, this may present with weight loss, altered bowel habit, or even metastasis to the liver or elsewhere. FAP can also develop 'silently' in some individuals, giving few or no signs until it has developed into advanced colorectal cancer.
Because familial polyposis develops very gradually over years, and can also manifest in an 'attenuated' form even more gradually, polyps resulting from FAP can lead to cancer developing at any point from adolescence to old age.
Depending on the nature of the defect in the APC gene, and whether it is the full or attenuated form, familial polyposis may manifest as polyps in colon or in the duodenal tract, or in any combination of these. Therefore, an absence of polyps in, for example, the rectum, may not of itself be sufficient to confirm absence of polyps. It may be necessary to consider and visually examine other possible parts of the intestinal tract. Colonoscopy is preferred over sigmoidoscopy for this, as it provides better observation of the common right-side location of polyps.

The genetic determinant in familial polyposis may also predispose carriers to other malignancies, e.g., of the duodenum and stomach (particularly ampullary adenocarcinoma). Other signs that may point to FAP are the development of Gardner fibromas and desmoid tumors (benign skin tumors that may be apparent before other signs of FAP), pigmented lesions of the retina ("CHRPE—congenital hypertrophy of the retinal pigment epithelium"), jaw cysts, sebaceous cysts, and osteomata (benign bone tumors). The combination of polyposis, osteomas, fibromas and sebaceous cysts is termed Gardner's syndrome (with or without abnormal scarring).
Genetics
Familial adenomatous polyposis can have different inheritance patterns and different genetic causes. When this condition results from mutations in the APC gene, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene is sufficient to cause the disorder. The incidence of malignancy in these cases approaches 100%. In most cases, an affected person has one parent with the condition.
APC gene mutation variants
The APC is a tumour suppressor gene responsible for the production of adenomatous polyposis coli (APC), a large multifunction tumour-suppressing protein which acts as a "gatekeeper" to prevent development of tumours. (APC regulates β-catenin, a protein that plays a crucial role in cell communication, signalling, growth, and controlled destruction, but which left uncontrolled also gives rise to numerous cancers). A flaw in the APC gene means APC is not as effective as it should be, and over time it is likely that some cells that should have been controlled by APC will not be, and will instead continue to develop and become cancerous. In familiar polyposis they usually manifest as polyps—small abnormalities on the surface of the intestinal tract.
Although the polyps are inherently benign, the first step of the two-hit hypothesis has already taken place: the inherited APC mutation. Often, the remaining "normal" allele is mutated or deleted, accelerating generation of polyps. Further mutations (e.g., in p53 or kRAS) to APC-mutated cells are much more likely to lead to cancer than they would in non-mutated epithelial cells.
The normal function of the APC gene product is still being investigated; it is present in both the cell nucleus and the membrane. The canonical tumor-suppressor function of APC is suppression of β-catenin, but other tumor-suppressor functions of APC may be related to cell adherence and cytoskeleton organization.
Mutation of APC also occurs commonly in incident cases of colorectal carcinoma, emphasizing its importance in this form of cancer.
MUTYH gene mutation variants
MUTYH encodes DNA repair enzyme MYH glycosylase. During normal cellular activities, guanine sometimes becomes altered by oxygen, which causes it to pair with adenine instead of cytosine. MYH glycosylase fixes these mistakes by base excision repair, such that mutations do not accumulate in the DNA and lead to tumor formation. When MYH glycosylase does not function correctly, DNA errors may accrue to initiate tumorigenesis with a clinical presentation similar to that in patients with APC mutations.
Mutations in the MUTYH gene are inherited in an autosomal recessive pattern, which means two copies of the gene must be altered for a person to be affected by the disorder. Most often, the parents of a child with an autosomal recessive disorder are not affected but are carriers of one copy of the altered gene.
Animal models
The "ApcMin" mouse model was described in 1990 and carries an Apc allele with a stop codon at position 850. Heterozygosity for this mutation results in a fully penetrant phenotype on most genetic backgrounds, with mice on a sensitive background developing over 100 tumors in the intestinal tract. The number and location of the intestinal tumors are modified by unlinked genes. Many other models have since appeared, including a model of attenuated FAP (the 1638N model) and several conditional mutants that allow for tissue-specific or temporal ablation of gene function. For more information see mouse models of colorectal and intestinal cancer.
In 2007, the "ApcPirc" rat model was isolated with a stop codon at position 1137. In contrast to the mouse models where >90% of tumors form in the small intestine, the Pirc rat forms tumors preferentially (>60%) in the large intestine, similar to the human clinical presentation.
Diagnosis

Making the diagnosis of FAP before the development of colon cancer is important not just for the individual, but also for the sake of other family members who may be affected. Two diagnostic methods exist:
- Colonoscopy is the usual diagnostic test of choice as it favours the common right-side location of polyps better than sigmoidoscopy if the mutation is attenuated FAP, and can confirm or allow (a) the actual clinical presentation and any change to the condition, of the 'at risk' individual, (b) quantification of polyps throughout the colon, (c) a histologic diagnosis (cell/cancer type detection) and (d) where polyps exist, it can suggest whether outpatient excision (removal) is viable or surgery is recommended. Barium enema and virtual colonoscopy (a form of medical imaging) can also be used to suggest the diagnosis of FAP.
- Genetic testing provides the ultimate diagnosis in 95% of cases; genetic counseling is usually needed in families where FAP has been diagnosed. Testing may also aid in the diagnosis of borderline cases in families that are otherwise known to p34.3 and p32.1 (1p34.3–p32.1). Testing can only show if an individual is susceptible to FAP or rule it out (i.e., whether or not they inherited the defective APC gene). It cannot determine the actual condition of a patient; this can only be found by direct physical examination.
NCBI states that physicians must ensure they understand the "risks, benefits, and limitations" of any genetic test done since in 1997 "for almost one-third of individuals assessed for FAP, the physician misinterpreted the test results".
Once the diagnosis of FAP is made, close colonoscopic surveillance with polypectomy is required.
Prenatal testing is possible if a disease-causing mutation is identified in an affected family member; however, prenatal testing for typically adult-onset disorders is uncommon and requires careful genetic counseling.
Ultrasound of the abdomen and blood tests evaluating liver function are often performed to rule out metastasis to the liver.
Management

Because of the way familial polyposis develops, it is possible to have the genetic condition, and therefore be at risk, but have no polyps or issues so far. Therefore, an individual may be diagnosed "at risk of" FAP, and require routine monitoring, but not (yet) actually have FAP (i.e., carries a defective gene but as yet appears not to have any actual medical issue as a result of this). Clinical management can cover several areas:
- Identifying those individuals who could be at risk of FAP: usually from family medical history or genetic testing
- Diagnosis (confirming whether they have FAP)—this can be done either by genetic testing, which is definitive or by visually checking the intestinal tract itself.
- It is important to note that visual examination, or monitoring, cannot 'clear' a person of risk. It can only say what their condition is at the time. If at any point in their life the person develops numerous polyps, this would tend to suggest a diagnosis of FAP. (Absence of polyps does not 'clear' a person, as polyps can develop later in life; also a few polyps over time are not that uncommon in people without FAP. However a substantial number or a profusion of polyps would generally tend to suggest a diagnosis of FAP, and histopathology to determine whether or not any polyps are cancerous.)
- Screening/monitoring programs involve visually examining the intestinal tract to check its healthy condition. It is undertaken as a routine matter every few years where there is cause for concern when either (a) a genetic test has confirmed the risk or (b) a genetic test has not been undertaken for any reason so the actual risk is unknown. Screening and monitoring allow polyposis to be detected visually before it can become life-threatening.
- Treatment, typically surgery of some kind, is involved if polyposis has led to a large number of polyps, or a significant risk of cancer, or actual cancer.
Family history
NCBI states that "Although most individuals diagnosed with an APC-associated polyposis condition have an affected parent, the family history may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent." In addition around 20% of cases are a de novo mutation, and of those with an apparent de novo APC mutation (i.e. no known family history) 20% have somatic mosaicism. Asymptomatic individuals (and therefore asymptomatic family members) are also known to exist.
Monitoring
Monitoring involves the provision of outpatient colonoscopy, and occasionally upper gastric tract esophagogastroduodenoscopy (EGD, to search for premalignant gastric or duodenal cancers), typically once every 1–3 years, and/or a genetic blood test to definitively confirm or deny susceptibility. A small number of polyps can often be excised (removed) during the procedure if found, but if there are more severe signs or numbers, inpatient surgery may be required.
NCBI states that when an individual is identified as having FAP, or the mutations resulting in FAP: "It is appropriate to evaluate the parents of an affected individual (a) with molecular genetic testing of APC if the disease-causing mutation is known in the proband [person first identified with the condition] or (b) for clinical manifestations of APC-associated polyposis conditions".
Treatment
Treatment for FAP depends on the genotype. Most individuals with the APC mutation will develop colon cancer by the age of 40, although the less-common attenuated version typically manifests later in life (40–70). Accordingly, in many cases, prophylactic surgery may be recommended before the age of 25, or upon detection if actively monitored. There are several surgical options that involve the removal of either the colon or both the colon and rectum.
- Rectum involved: the rectum and part or all of the colon are removed. The patient may require an ileostomy (permanent stoma where stool goes into a bag on the abdomen) or have an ileo-anal pouch reconstruction. The decision to remove the rectum depends on the number of polyps in the rectum as well as the family history. If the rectum has few polyps, the colon is partly or fully removed and the small bowel (ileum) can be directly connected to the rectum instead (ileorectal anastomosis).
- Rectum not involved: the portion of the colon manifesting polyps can be removed and the ends 'rejoined' (partial colectomy), a surgery that has a substantial healing time but leaves quality of life largely intact.
Prophylactic colectomy is indicated if more than a hundred polyps are present, if there are severely dysplastic polyps, or if multiple polyps larger than 1 cm are present.
Treatment for the two milder forms of FAP may be substantially different from the more usual variant, as the number of polyps is far fewer, allowing more options.
Various medications are being investigated for slowing malignant degeneration of polyps, most prominently the non-steroidal anti-inflammatory drugs (NSAIDs). NSAIDS have been shown to significantly decrease the number of polyps but do not usually alter management since there are still too many polyps to be followed and treated endoscopically. The drug eflornithine, an inhibitor of ornithine decarboxylase typically used to treat trypanosomiasis, is being investigated as a potential preventive medication in combination with the NSAID celecoxib for treatment of FAP. Another investigational agent is sulindac, also used in combination with NSAIDs.
Prognosis
Prior to reaching the advanced stages of colorectal cancer, the polyps are confined to the inner wall and thickness of the intestinal tract and do not metastasize or 'spread'. So provided FAP is detected and controlled either at the pre-cancerous stage or when any cancerous polyps are still internal to the intestinal tract, surgery has a very high success rate of preventing or removing cancer, without recurrence, since the locations giving rise to cancer are physically removed in toto by the surgery.
Following surgery, if a partial colectomy has been performed, colonoscopic surveillance of the remaining colon is necessary as the individual still has a risk of developing colon cancer. However, if this happened, it would be a fresh incident from polyps developing anew in the unremoved part of the colon subsequent to surgery, rather than a return or metastasis of any cancer removed by the original surgery.
Epidemiology
The incidence of the mutation is between 1 in 10,000 and 1 in 15,000 births. By age 35 years, 95% of individuals with FAP (>100 adenomas) have polyps. Without colectomy, colon cancer is virtually inevitable. The mean age of colon cancer in untreated individuals is 39 years (range 34–43 years).
Attenuated FAP arises when APC is defective but still somewhat functional. As a result, it retains part of its ability to suppress polyps. Therefore, attenuated FAP manifests as colorectal cancer unusually late (age 40–70, average=55), and typically with few, or at least far fewer polyps (typically 30), than the more usual version of FAP, at an age when FAP is no longer considered much of a likelihood or risk according to usual FAP epidemiology.
Comparison of FAP variants
This table compares the different subtypes of FAP:
| Item | FAP | Attenuated FAP | MUTYH Associated FAP |
| Gene | APC | APC | MUTYH |
| Typical polyp manifestation | Hundreds / thousands | Under 100 (0–470, typ. 30), sometimes flat rather than polypoid morphology, and more proximal to the splenic flexure. In a study of 120 individuals 37% (N=44) had <10 polyps; 3 of these 44 had colorectal cancer]. Gastric fundic polyps and duodenal adenomas are also seen. Therefore, polyps and cancers may manifest in the upper portion of the colon or upper gastrointestinal tract rather than the usual locations | ? |
| Typical core diagnostic criteria | (a) 100+ polyps and age under 40, OR (b) polyps and FAP in a relative | Not settled as yet. (a) no family history of 100+ polyps before age 30 PLUS ONE OF 10–99 polyps / 100+ polyps and aged over 35 to 40 / colorectal cancer before age 60 and relatives with multiple adenomatous polyps, OR (b) Family history of 10 to 99 adenomas diagnosed after age 30 years | ? |
| Age at which polyps manifest | 7–36 (typ. 16), rapidly increasing thereafter | ? | ? |
| Colorectal cancer risk (penetrance) and age if untreated | "inevitable.. virtually 100%": 7% by age 21, 87% by age 45, 93% by age 50. Typical ages: 34–43 (avg.39) | "Lower.. less well known.. estimated 70% by age 80". Sovaria states as at 1998, "average age at CRC diagnosis is ~58 years" | ? |
| Variability | Inter- and intrafamilial phenotypic variability are common | See FAP | ? |
| Possible non-colon manifestations | "polyps of the gastric fundus and duodenum, osteomas, dental anomalies, congenital hypertrophy of the retinal pigment epithelium (CHRPE), soft tissue tumors, desmoid tumors, and associated cancers" | As for FAP but "CHRPE and desmoid tumors are rare" and also thyroid cancer is added. | ? |
| Other lifetime risks | "Small bowel [duodenum or periampulla] carcinoma 4–12% [distal to duodenum] Rare; Pancreas Adenocarcinoma ~1%; Papillary thyroid carcinoma 1–2%; CNS [typ. medulloblastoma] <1%; Liver hepatoblastoma 1.6%; Bile ducts adenocarcinoma Low but increased; Stomach adenocarcinoma <1% in Western cultures." | ? | ? |
| Inheritance | "inherited in an autosomal dominant manner. Approximately 75%-80% of individuals with APC-associated polyposis conditions have an affected parent. Offspring of an affected individual are at a 50% risk of inheriting the disease-causing mutation" | Same as FAP | Different—recessive (requires 2 parents to be carriers) |
| Genetic overview and genetic detection | "Full gene sequencing of all APC exons and intron-exon boundaries appears to be the most accurate clinical test available. Most APC mutations are nonsense or frameshift mutations that cause premature truncation of the APC protein.. The likelihood of detecting an APC mutation is highly dependent on the severity of colonic polyposis and on the family history.. ◦Approximately 20% of individuals with an apparent de novo APC mutation.. The markers used for linkage analysis of APC-associated polyposis conditions are highly informative and very tightly linked to the APC locus; thus, they can be used with greater than 98% accuracy in more than 95% of families with an APC-associated polyposis condition. Linkage testing is not possible for families with a single affected individual, a situation that often occurs when an individual has a de novo gene mutation and no affected offspring.. If no disease causing APC mutation is found, molecular genetic testing of MUTYH (see Differential Diagnosis) should be considered." | "Fewer than 30% of individuals with attenuated phenotypes are expected to have an identifiable APC mutation" (see also details under FAP) | ? |
| Genotype-Phenotype [Core condition] | Most frequent APC mutation is at codon 1309 and lead to a high number of polyps at an early age (~20). Profuse polyposis (avg=5000) reported with mutations in codons 1250–1464. Most partial and whole APC deletions are associated with 100–2000 colonic adenomas, although attenuated FAP has been seen. Sample typical onset ages: between codon 168 and 1580 (excluding 1309) = 30 years, 5' of codon 168 and 3' of codon 1580 = 52 years. | Attenuated FAP is associated with mutations (typically truncating) in the 5' part of the gene (codons 1–177), exon 9, and the distal 3' end of the gene; interstitial deletions of chromosome 5q22 that include APC; partial and whole-gene deletions; and somatic mosaicism for APC mutations that are generally associated with classic FAP. Sovaria states attenuated FAP is "caused by mutations in three distinct regions of the APC gene—the 5′ end in the region spanning exons 4 and 5, exon 9, and the extreme 3′ end. Phenotypic expression in these three groups of kindreds is variable but is definitely milder than that in classical FAP" and that rectal polyps are rare in attenuated FAP but not yet confirmed whether this also means rectal cancer risk is lower as well. | ? |
| Genotype–Phenotype [Other extra-colonic conditions] | Prominent extracolonic manifestations often correlate (though not completely) with more distal APC mutations. General study of FAP plus extracolonic symptoms showed: mutations in codons 1395–1493 has significantly higher rates of desmoid tumors, osteomas, and epidermoid cysts than those with mutations in codons 177–452; mutations in codons 1395–1493 have significantly higher rates of desmoid tumors and osteomas than those with mutations in codons 457–1309; no individuals with mutations in codons 177–452 developed osteomas or periampullary cancers; only individuals with mutations in codons 457–1309 developed hepatoblastoma and/or brain tumors. Duodenal adenomas: Fourfold increased risk with mutations between codons 976 and 1067. Desmoid tumors: mutations 3' to codon 1399 were associated with desmoid tumor development with an odds ratio of 4.37; desmoid tumors in 20% of individuals with mutations 5' to codon 1444, 49% of individuals with mutations 3' to codon 1444, and 61% of individuals with mutations in codons 1445–1580; several families with severe desmoid tumors had mutations at the extreme 3' end; consistent association of desmoid tumors with mutations distal to codon 1444. CHRPE is associated with: mutations between codons 311 and 1444; whole APC gene deletions. Thyroid cancer and FAP: In 24 individuals, the majority of mutations identified were 5' to codon 1220 [Cetta et al. 2000]; 9 of 12 individuals had APC mutations identified proximal to the mutation cluster region (codons 1286–1513). General review of the literature (to August 2006): revealed 89 submicroscopic APC deletions (42 partial and 47 whole-gene deletions). Extracolonic findings were seen in 36% of cases, with no significant differences in those with partial vs. whole-gene deletions. | ? | ? |
| Prevalence | "2.29 to 3.2 per 100,000 individuals.. APC-associated polyposis conditions historically accounted for about 0.5% of all colorectal cancers; this figure is declining as more at-risk family members undergo successful treatment following early polyp detection and prophylactic colectomy." | "Likely underdiagnosed, given the lower number of colonic polyps and lower risk for colorectal cancer compared to classic FAP" | ? |
| Treatment of manifestations | Classic FAP: "Colectomy is recommended after adenomas emerge; colectomy may be delayed depending on the size and number of adenomatous polyps. Colectomy is usually advised when more than 20 or 30 adenomas or multiple adenomas with advanced histology have developed" | "Colectomy may be necessary, but in approximately one third of individuals the colonic polyps are limited enough in number that surveillance with periodic colonoscopic polypectomy is sufficient" | ? |
| Surveillance (monitoring) activities once risk is established | "Sigmoidoscopy or colonoscopy every 1–2 years, beginning at age ten to 12 years; colonoscopy, once polyps are detected; annual colonoscopy, if colectomy is delayed more than a year after polyps emerge (Age ten to 20 years with certain milder symptoms, delay in colectomy may be considered); Esophagogastroduodenoscopy (EGD) by age 25 years or prior to colectomy and repeated every 1–3 years; in some cases, endoscopic retrograde cholangiopancreatography (ERCP) to evaluate for adenomas of the common bile duct; small-bowel imaging when duodenal adenomas are detected or prior to colectomy, repeated every 1–3 years depending on findings; screening for hepatoblastoma (optimal interval unknown, one paper recommends "at least every three months"); annual physical examination, including evaluation for extraintestinal manifestations, and palpation of the thyroid with consideration of follow-up ultrasound examination and fine-needle aspiration if thyroid nodules are present" | "Colonoscopy every two to three years, beginning at age 18 to 20 years; esophagogastroduodenoscopy (EGD) beginning by age 25 years or prior to colectomy and repeated every 1–3 years; in some cases, endoscopic retrograde cholangiopancreatography (ERCP) may be necessary to evaluate for adenomas of the common bile duct; annual physical examination with palpation of the thyroid with consideration of follow-up ultrasound examination and fine-needle aspiration if thyroid nodules are present. Colectomy usually advised when more than 20 or 30 adenomas or multiple adenomas with advanced histology have developed." Sovaria states as at 1998 that "colonoscopy, as opposed to sigmoidoscopy, should be advised for endoscopic surveillance, because of the right-side location of colorectal adenomas; UGI endoscopic surveillance is warranted in an attempt to detect premalignant gastric or duodenal tumors; individuals affected with [attenuated FAP] may require total colectomy with ileo-rectal anastomosis only when prophylactic colectomy is advised" | ? |
| Decision to monitor | "Early recognition may allow for timely intervention and improved final outcome; thus, surveillance of asymptomatic, at-risk children for early manifestations is appropriate; genetic testing is more cost effective than sigmoidoscopy in determining who in the family is affected; individuals diagnosed with APC-associated polyposis conditions as a result of having an affected relative have a significantly greater life expectancy than those individuals diagnosed on the basis of symptoms.. As colon monitoring for those at risk for classic FAP begins as early as age ten to 12 years, molecular genetic testing is generally offered to children at risk for classic FAP by age ten years. Genetic testing at birth may also be warranted, as some parents and pediatricians may consider hepatoblastoma screening from infancy to age five years in affected offspring.. No evidence points to an optimal age at which to begin screening." | See FAP. Also "Colon screening for those with attenuated FAP begins at age 18 to 20 years; thus, molecular genetic testing should be offered to those at risk for attenuated FAP at approximately age 18 years." | ? |
| Inheritance and implications of confirmed diagnosis for other close relatives | APC-associated polyposis conditions are inherited in an autosomal dominant manner. Approximately 20–25% have the altered gene as the result of a de novo gene mutation. Little or no evidence of maternal/paternal bias, or effect related to advanced paternal age, in de novo mutations. Siblings have classic 50% risk of sharing the condition if inherited and not de novo and a "low" but slightly higher risk than general if de novo, therefore genetic testing should be offered. Offspring each have a 50% chance of inheritance. Other family members are at risk if their parents share the same mutation. Germline mosaicism has been documented in asymptomatic cases. Prenatal testing is possible via fetal extracted DNA. | See FAP | ? |
Polyposis registries
Because of the genetic nature of FAP, polyposis registries have been developed around the world. The purpose of these registries is to increase knowledge about the transmissibility of FAP, but also to document, track, and notify family members of affected individuals. One study has shown that the use of a registry to notify family members (call-ups) significantly reduced mortality when compared with probands. The St. Mark's polyposis registry is the oldest in the world, started in 1924, and many other polyposis registries now exist.
See also
Further reading
- Jasperson, Kory W.; Patel, Swati G.; Ahnen, Dennis J. (2 February 2017). "APC-Associated Polyposis Conditions". In Adam MP; Ardinger HH; Pagon RA; Wallace SE (eds.). GeneReviews. Seattle WA: University of Washington. PMID 20301519. NBK1345. — full clinical summary of FAP and attenuated FAP, including lifetime risks, epidemiology etc.
- Familial adenomatous polyposis at NLM Genetics Home Reference
- Familial Adenomatous Polyposis—eMedicine Gastroenterology
- Colon, Polyposis Syndromes
- National Cancer Institute: Genetics of Colorectal Cancer information summary