
| Gerstmann–Sträussler–Scheinker syndrome | |
|---|---|
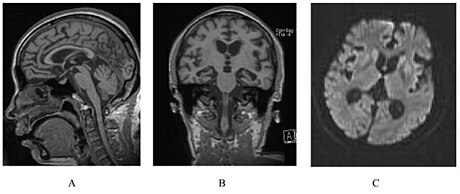 | |
| A person with inherited prion disease has cerebellar atrophy. This is quite typical of GSS. | |
| Specialty |
Neurology |
| Symptoms | difficulty speaking, developing dementia, memory loss, vision loss. |
| Causes | prion |
| Prognosis | Universally fatal, life expectancy is typically 5-6 years from diagnosis |
Gerstmann–Sträussler–Scheinker syndrome (GSS) is an extremely rare, usually familial, fatal neurodegenerative disease that affects patients from 20 to 60 years in age. It is exclusively heritable, and is found in only a few families all over the world. It is, however, classified with the transmissible spongiform encephalopathies (TSE) due to the causative role played by PRNP, the human prion protein. GSS was first reported by the Austrian physicians Josef Gerstmann, Ernst Sträussler and Ilya Scheinker in 1936.
Familial cases are associated with autosomal-dominant inheritance.
Certain symptoms are common to GSS, such as progressive ataxia, pyramidal signs, and dementia; they worsen as the disease progresses.
Symptoms and signs
Symptoms start with slowly developing dysarthria (difficulty speaking) and cerebellar truncal ataxia (unsteadiness) and then the progressive dementia becomes more evident. In the early stages of GSS, people with the condition may also exhibit clumsiness and experience difficulty walking. As the condition progresses, symptoms of ataxia become more pronounced. Loss of memory can be the first symptom of GSS. Extrapyramidal and pyramidal symptoms and signs may occur and the disease may mimic spinocerebellar ataxias in the beginning stages. Myoclonus (spasmodic muscle contraction) is less frequently seen than in Creutzfeldt–Jakob disease. Many patients also exhibit nystagmus (involuntary movement of the eyes), visual disturbances, and even blindness or deafness. The neuropathological findings of GSS include widespread deposition of amyloid plaques composed of abnormally folded prion protein.
Four clinical phenotypes are recognized: typical GSS, GSS with areflexia and paresthesia, pure dementia GSS and Creutzfeldt-Jakob disease-like GSS.
Causes
GSS is part of a group of diseases called transmissible spongiform encephalopathies. These diseases are caused by prions, which are a class of pathogenic proteins that are resistant to proteases. These prions then form clusters in the brain, which are responsible for the neurodegenerative effects seen in patients.
The P102L mutation, which causes a substitution of proline to a leucine in codon 102, has been found in the prion protein gene (PRNP, on chromosome 20) of most affected individuals. Therefore, it appears this genetic change is usually required for the development of the disease.
Diagnosis
GSS can be identified through genetic testing. Testing for GSS involves a blood and DNA examination in order to attempt to detect the mutated gene at certain codons. If the genetic mutation is present, the patient will eventually develop GSS.
Treatment
There is no cure for GSS, nor is there any known treatment to slow the progression of the disease. Therapies and medication are aimed at treating or slowing down the effects of the symptoms. The goal of these treatments is to try to improve the patient's quality of life as much as possible. There is some ongoing research to find a cure, with one of the most prominent examples being the PRN100 monoclonal antibody.
Prognosis
GSS is a disease that progresses slowly, lasting roughly 2–10 years, with an average of approximately five years. The disease ultimately results in death, most commonly from the patient either going into a coma, or from a secondary infection due to the patient's loss of bodily functions.
Research
Prion diseases, also called transmissible spongiform encephalopathies (TSEs), are neurodegenerative diseases of the brain thought to be caused by a protein that converts to an abnormal form called a prion. GSS is a very rare TSE, making its genetic origin nearly impossible to determine. It is also challenging to find any patients with GSS, as the disease tends to be underreported, due to its clinical similarity to other diseases, and has been found in only a few countries. In 1989, the first mutation of the prion protein gene was identified in a GSS family. The largest of these families affected by GSS is the Indiana Kindred, spanning over 8 generations, and includes over 3,000 people, with 57 individuals known to be affected. GSS was later realized to have many different gene mutation types, varying in symptom severity, timing and progression. Doctors in different parts of the world are in the process of uncovering more generations and families who have the mutation.
External links
- Gerstmann–Sträussler–Scheinker syndrome, MedicineNet.com