
| Glycogen storage disease | |
|---|---|
| Other names | Glycogenosis, dextrinosis |
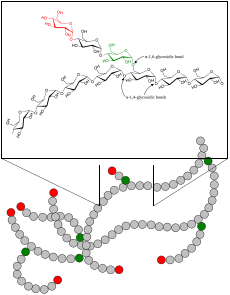 | |
| Glycogen | |
| Specialty |
Endocrinology |
A glycogen storage disease (GSD, also glycogenosis and dextrinosis) is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.
GSD has two classes of cause: genetic and acquired. Genetic GSD is caused by any inborn error of carbohydrate metabolism (genetically defective enzymes or transport proteins) involved in these processes. In livestock, acquired GSD is caused by intoxication with the alkaloid castanospermine.
However, not every inborn error of carbohydrate metabolism has been assigned a GSD number, even if it is known to affect the muscles or liver. For example, Phosphoglycerate Kinase Deficiency (gene PGK1) has a myopathic form.
Also, Fanconi-Bickel syndrome (gene SLC2A2) and Danon disease (gene LAMP2) were declassed as GSDs due to being defects of transport proteins rather than enzymes; however, GSD-1 subtypes b, c, and d are due to defects of transport proteins (genes SLC37A4, SLC17A3) yet are still considered GSDs.
Phosphoglucomutase deficiency (gene PGM1) was declassed as a GSD due to it also affecting the formation of N-glycans; however, as it affects both glycogenolysis and glycosylation, it has been suggested that it should re-designated as GSD-XIV.
(See Inborn Errors of Carbohydrate Metabolism for a full list of inherited diseases that affect glycogen synthesis, glycogen breakdown, or glucose breakdown.)
Types
| Type (Eponym) |
Enzyme deficiency (Gene) |
Incidence (births) |
Hypo- glycemia? |
Hepato- megaly? |
Hyper- lipidemia? |
Muscle symptoms | Development/ prognosis | Other symptoms |
|---|---|---|---|---|---|---|---|---|
| GSD 0 |
Glycogen synthase (GYS2) |
? | Yes | No | No | Occasional muscle cramping | Growth failure in some cases | |
|
GSD I / GSD 1 (von Gierke's disease) |
Glucose-6-phosphatase / Glucose-6-phosphate translocase (G6PC / SLC37A4 /SLC17A3) |
1 in 50,000 – 100,000 | Yes | Yes | Yes | None | Growth failure | Lactic acidosis, hyperuricemia |
|
GSD II / GSD 2 (Pompe disease, formerly GSD-IIa) Danon disease (formerly GSD-IIb) |
Acid alpha-glucosidase
(GAA) Lysosome-associated membrane protein 2 (LAMP2) |
Pompe disease is 1 in 13,000. | No | Yes | No |
Muscle weakness, exercise intolerance, abnormal glycogen accumulation in muscle biopsy.
The symptoms of both Pompe and Danon diseases are very similar due to a defect in lysosomes. However, in Danon disease, some show abnormal glycogen accumulation, but not all. |
Progressive proximal skeletal muscle weakness with varied timeline to threshold of functional limitation (early childhood to adulthood). Approximately 15% of the Pompe population is classified as infantile Pompe which is typically deadly within the first year if untreated. | Heart failure (infantile), respiratory difficulty (due to muscle weakness) |
|
GSD III / GSD 3 (Cori's disease or Forbes' disease) |
Glycogen debranching enzyme (AGL) |
1 in 100,000 | Yes | Yes | Yes | Myopathy |
myogenic hyperuricemia |
|
|
GSD IV / GSD 4 (Andersen's disease) |
Glycogen branching enzyme (GBE1) |
1 in 500,000 | No | Yes, also cirrhosis |
No | Myopathy and dilated cardiomyopathy | Failure to thrive, death at age ~5 years | |
|
GSD V / GSD 5 (McArdle's disease) |
Muscle glycogen phosphorylase (PYGM) |
1 in 100,000 – 500,000 | No | No | No | Exercise-induced muscle fatigue and cramps. Rhabdomyolysis possible. Some have hypertrophic calf muscles. | Renal failure by myoglobinuria, second wind phenomenon, inappropriate rapid heart rate (sinus tachycardia) response to exercise, myogenic hyperuricemia | |
|
GSD VI / GSD 6 (Hers' disease) |
Liver glycogen phosphorylase (PYGL) |
1 in 65,000 – 85,000 | Yes | Yes | Yes | None | initially benign, developmental delay follows. | |
|
GSD VII / GSD 7 (Tarui's disease) |
Muscle phosphofructokinase (PFKM) |
1 in 1,000,000 | No | No | No | Exercise-induced muscle cramps and weakness | developmental delay | In some haemolytic anaemia,
myogenic hyperuricemia |
| GSD IX / GSD 9 |
Phosphorylase kinase (PHKA2 / PHKB / PHKG2 / PHKA1) |
? | Yes | Yes | Yes | None | Delayed motor development, Developmental delay | |
| GSD X / GSD 10 | Muscle Phosphoglycerate mutase(PGAM2) | ? | ? | ? | ? | Exercise-induced muscle cramps and weakness | Myoglobinuria | |
| GSD XI / GSD 11 |
Muscle lactate dehydrogenase (LDHA) |
? | ? | ? | ? | |||
|
Fanconi-Bickel syndrome formerly GSD XI / GSD 11, no longer considered a GSD |
Glucose transporter (GLUT2) |
? | Yes |
Yes |
No | None | ||
| GSD XII / GSD 12 (Aldolase A deficiency) |
Aldolase A (ALDOA) |
? | No | In some | No | Exercise intolerance, cramps. In some Rhabdomyolysis. | Hemolytic anemia and other symptoms | |
| GSD XIII / GSD 13 |
β-enolase (ENO3) |
? | No | ? | No | Exercise intolerance, cramps | Increasing intensity of myalgias over decades | Serum CK: Episodic elevations; Reduced with rest |
| CDG1T (formally GSD XIV / GSD 14) | Phosphoglucomutase-1(PGM1) | ? | Episodic | ? | No | Two forms: exclusively myopathic and multi-system (including muscles). myopathy (including exercise-related fatigue, exercise intolerance, muscle weakness). Muscle biopsy shows glycogen accumulation. |
Short stature, some have developmental delay, and rarely delayed puberty. | Highly variable phenotype and severity. Commonly elevated serum CK, abnormal serum transferrin (loss of complete N-glycans), short stature, cleft palate, bifid uvula, and hepatopathy. Second Wind phenomenon in some but not all |
| GSD XV / GSD 15 |
Glycogenin-1 (GYG1) |
Rare | No | No | No | Muscle atrophy, exercise intolerance, muscle biopsy shows abnormal glycogen depletion and marked proliferation of slow-twitch (type 1/oxidative) muscle fibres and mitochondrial proliferation. | Slowly progressive weakness over decades | Arrhythmia, biopsy of heart showed abnormal glycogen deposits (different from polyglucosan bodies) in cardiomyocytes. |
Remarks:
- Some GSDs have different forms, e.g. infantile, juvenile, adult (late-onset).
- Some GSDs have different subtypes, e.g. GSD1a / GSD1b, GSD9A1 / GSD9A2 / GSD9B / GSD9C / GSD9D.
- GSD type 0: Although glycogen synthase deficiency does not result in storage of extra glycogen in the liver, it is often classified with the GSDs as type 0 because it is another defect of glycogen storage and can cause similar problems.
- GSD type VIII (GSD 8): In the past, Liver Phosphorylase-b Kinase Deficiency was considered a distinct condition, however it is has been classified with GSD type VI and GSD IXa1; it has been described as X-linked recessive inherited. GSD IX has become the dominant classification for this disease, grouped with the other isoenzymes of Phosphorylase-b Kinase Deficiency.
- GSD type XI (GSD 11): Fanconi-Bickel syndrome (GLUT2 deficiency), hepatorenal glycogenosis with renal Fanconi syndrome, no longer considered a glycogen storage disease, but a defect of glucose transport. The designation of GSD type XI (GSD 11) has been repurposed for muscle lactate dehydrogenase deficiency (LDHA).
- GSD type XIV (GSD 14): Now classed as Congenital disorder of glycosylation type 1T (CDG1T), affects the phosphoglucomutase enzyme (gene PGM1).Phosphoglucomutase 1 Deficiency is both a glycogenosis and a congenital disorder of glycosylation. Individuals with the disease have both a glycolytic block as muscle glycogen cannot be broken down, as well as abnormal serum transferrin (loss of complete N-glycans).
- Lafora disease is considered a complex neurodegenerative disease and also a glycogen metabolism disorder.
- Polyglucosan Storage Myopathies are associated with defective glycogen metabolism
- (Not McArdle Disease, same gene but different symptoms) Myophosphorylase-a activity impaired: Autosomal dominant mutation on PYGM gene. AMP-independent myophosphorylase activity impaired, whereas the AMP-dependent activity was preserved. No exercise intolerance. Adult-onset muscle weakness. Accumulation of the intermediate filament desmin in the myofibers of the patients. Myophosphorylase comes in two forms: form 'a' is phosphorylated by phosporylase kinase, form 'b' is not phosphorylated. Both forms have two conformational states: active (R or relaxed) and inactive (T or tense). When either form 'a' or 'b' are in the active state, then the enzyme converts glycogen into glucose-1-phosphate. Myophosphorylase-b is allosterically activated by AMP being in larger concentration than ATP and/or glucose-6-phosphate. (See Glycogen phosphorylase§Regulation).
- Unknown glycogenosis related to dystrophy gene deletion: patient has a previously undescribed myopathy associated with both Becker muscular dystrophy and a glycogen storage disorder of unknown aetiology.
Diagnosis
Methods to diagnose glycogen storage diseases include history and physical examination for associated symptoms, blood tests for associated metabolic disturbances, and genetic testing for suspected mutations. It may also include a non-ischemic forearm test, exercise stress test, or 12-minute walk test (12MWT).
Treatment
Treatment is dependent on the type of glycogen storage disease. Von Gierke disease (GSD-I) is typically treated with frequent small meals of carbohydrates and cornstarch, called modified cornstarch therapy, to prevent low blood sugar, while other treatments may include allopurinol and human granulocyte colony stimulating factor.
Cori/Forbes disease (GSD-III) treatment may use modified cornstarch therapy, a high protein diet with a preference to complex carbohydrates. However, unlike GSD-I, gluconeogenesis is functional, so simple sugars (sucrose, fructose, and lactose) are not prohibited.
A ketogenic diet has demonstrated beneficial for McArdle disease (GSD-V) as ketones readily convert to Acetyl CoA for oxidative phosphorylation, whereas Free Fatty Acids take a few minutes to convert into Acetyl CoA.
For phosphoglucomutase deficiency (formerly GSD-XIV), D-galactose supplements and exercise training has shown favourable improvement of signs and symptoms. In terms of exercise training, some patients with phosphoglucomutase deficiency also experience "second wind."
For McArdle disease (GSD-V), regular aerobic exercise utilizing "second wind" to enable the muscles to become aerobically conditioned, as well as anaerobic exercise (strength training) that follows the activity adaptations so as not to cause muscle injury, helps to improve exercise intolerance symptoms and maintain overall health. Studies have shown that regular low-moderate aerobic exercise increases peak power output, increases peak oxygen uptake (VO2peak), lowers heart rate, and lowers serum CK in individuals with McArdle disease.
Regardless of whether the patient experiences symptoms of muscle pain, muscle fatigue, or cramping, the phenomenon of second wind having been achieved is demonstrable by the sign of an increased heart rate dropping while maintaining the same speed on the treadmill. Inactive patients experienced second wind, demonstrated through relief of typical symptoms and the sign of an increased heart rate dropping, while performing low-moderate aerobic exercise (walking or brisk walking).
Conversely, patients that were regularly active did not experience the typical symptoms during low-moderate aerobic exercise (walking or brisk walking), but still demonstrated second wind by the sign of an increased heart rate dropping. For the regularly active patients, it took more strenuous exercise (very brisk walking/jogging or bicycling) for them to experience both the typical symptoms and relief thereof, along with the sign of an increased heart rate dropping, demonstrating second wind.
In young children (<10 years old) with McArdle disease (GSD-V), it may be more difficult to detect the second wind phenomenon. They may show a normal heart rate, with normal or above normal peak cardio-respiratory capacity (VO2max). That said, patients with McArdle disease typically experience symptoms of exercise intolerance before the age of 10 years.
Tarui disease (GSD-VII) patients do not experience the "second wind" phenomenon; instead are said to be "out-of-wind." However, they can achieve sub-maximal benefit from lipid metabolism of free fatty acids during aerobic activity following a warm-up.
Epidemiology

Overall, according to a study in British Columbia, approximately 2.3 children per 100,000 births (1 in 43,000) have some form of glycogen storage disease. In the United States, they are estimated to occur in 1 per 20,000–25,000 births. Dutch incidence rate is estimated to be 1 per 40,000 births. While a Mexican incidence showed 6.78:1000 male newborns.
See also
External links
- IamGSD - International Association for Muscle Glycogen Storage Disease. A non-profit, patient-led international group encouraging efforts by research and medical professionals, national support groups and individual patients worldwide.
- IPA - International Pompe Association. (Pompe Disease is also known as GSD-II). A non-profit, federation of Pompe disease patient's groups world-wide. It seeks to coordinate activities and share experience and knowledge between different groups.
- EUROMAC - EUROMAC is a European registry of patients affected by McArdle Disease and other rare neuromuscular glycogenoses.
- CoRDS - Coordination of Rare Diseases at Sanford (CoRDS) is a centralized international patient registry for all rare diseases. They work with patient advocacy groups, including IamGSD, individuals and researchers.
- CORD - Canadian Organization for Rare Disorders (CORD) is a Canadian national network for organizations representing all those with rare disorders. CORD provides a strong common voice to advocate for health policy and a healthcare system that works for those with rare disorders.
- NORD - National Organization for Rare Disorders (NORD) is an American national non-profit patient advocacy organization that is dedicated to individuals with rare diseases and the organizations that serve them.
- EURODIS - Rare Diseases Europe (EURODIS) is a unique, non-profit alliance of over 700 rare disease patient organizations across Europe that work together to improve the lives of the 30 million people living with a rare disease in Europe.