
| Klinefelter syndrome | |
|---|---|
| Other names | XXY syndrome, Klinefelter's syndrome, Klinefelter-Reifenstein-Albright syndrome |
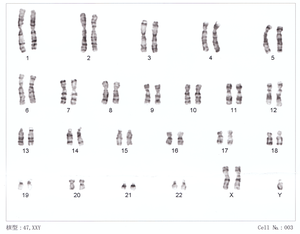 | |
| 47,XXY karyotype | |
| Pronunciation | |
| Specialty | Medical genetics |
| Symptoms | Above average height, weaker muscles, poor coordination, less body hair, breast growth, less interest in sex, infertility. |
| Complications | Infertility, autoimmune disorders, breast cancer, venous thromboembolic disease, osteoporosis |
| Usual onset | At fertilisation |
| Duration | Long term |
| Causes | Two or more X chromosomes in males |
| Risk factors | Older mother |
| Diagnostic method | Genetic testing (karyotype) |
| Prevention | None |
| Treatment | Physical therapy, speech and language therapy, counseling |
| Prognosis | Nearly normal life expectancy |
| Frequency | 1:500 to 1:1,000 males |
Klinefelter syndrome (KS), also known as 47,XXY, is an aneuploid genetic condition where a male has an additional copy of the X chromosome. The primary features are infertility and small, poorly functioning testicles. Usually, symptoms are subtle and subjects do not realize they are affected. Sometimes, symptoms are more evident and may include weaker muscles, greater height, poor motor coordination, less body hair, breast growth, and less interest in sex. Often, these symptoms are noticed only at puberty. Intelligence is usually average, but reading difficulties and problems with speech are more common.
Klinefelter syndrome occurs randomly. The extra X chromosome comes from the father and mother nearly equally. An older mother may have a slightly increased risk of a child with KS. The syndrome is defined by the presence of at least one extra X chromosome in addition to a Y chromosome yielding a total of 47 or more chromosomes rather than the usual 46. KS is diagnosed by the genetic test known as a karyotype.
While no cure is known, a number of treatments may help.Physical therapy, occupational therapy, speech and language therapy, counselling, and adjustments of teaching methods may be useful.Testosterone replacement may be used in those who have significantly lower levels. Enlarged breasts may be removed by surgery. Approximately half of affected males have a chance of fathering children with the help of assisted reproductive technology, but this is expensive and not risk free. XXY males have a ~15-fold higher risk of developing breast cancer than typical, but still lower than that of females. People with the condition have a nearly normal life expectancy.
Klinefelter syndrome is one of the most common chromosomal disorders, occurring in one to two per 1,000 live male births. It is named after American endocrinologist Harry Klinefelter, who identified the condition in the 1940s. In 1956, the extra X chromosome was identified as the cause. Mice can also have the XXY syndrome, making them a useful research model.
Signs and symptoms

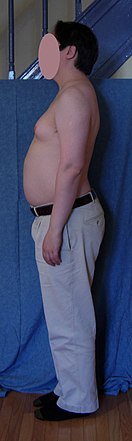
The primary features are infertility and small, poorly functioning testicles. Often, symptoms may be subtle and many people do not realize they are affected. Sometimes, symptoms are more prominent and may include weaker muscles, greater height, poor coordination, less body hair, breast growth, and low libido. Often, these symptoms are noticed only at puberty.
Prenatal
An estimated 60% of pregnancies with fetuses having Klinefelter syndrome abort.
Physical
As babies and children, XXY males may have weaker muscles and reduced strength. As they grow older, they tend to become taller than average. They may have less muscle control and coordination than other boys of their age.
During puberty, the physical traits of the syndrome become more evident; because these boys do not produce as much testosterone as other boys, they have a less muscular body, less facial and body hair, and broader hips. As teens, XXY males may develop breast tissue and also have weaker bones, and a lower energy level than other males.
By adulthood, XXY males look similar to males without the condition, although they are often taller. In adults, possible characteristics vary widely and include little to no sign of affectedness, a lanky, youthful build and facial appearance, or a rounded body type with some degree of gynecomastia (increased breast tissue). Gynecomastia is present in approximately a third of affected individuals, a slightly higher percentage than in the XY population. Approximately 10% of XXY males have gynecomastia noticeable enough that they may choose to have cosmetic surgery.
Affected males are often infertile, or have reduced fertility. Advanced reproductive assistance is sometimes possible. An estimated 50% of males with Klinefelter syndrome can produce sperm.
The term "hypogonadism" in XXY symptoms is often misinterpreted to mean "small testicles", when it instead means decreased testicular hormone/endocrine function. Because of (primary) hypogonadism, individuals often have a low serum testosterone level, but high serum follicle-stimulating hormone and luteinizing hormone levels, hypergonadotropic hypogonadism. Despite this misunderstanding of the term, however, XXY men may also have microorchidism (i.e., small testicles).
The testicles of affected males are usually less than 2 cm in length (and always shorter than 3.5 cm), 1 cm in width, and 4 ml in volume.
XXY males are more likely than other men to have certain health problems, such as autoimmune disorders, breast cancer, venous thromboembolic disease, and osteoporosis. In contrast to these potentially increased risks, rare X-linked recessive conditions are thought to occur less frequently in XXY males than in XY males, since these conditions are transmitted by genes on the X chromosome, and people with two X chromosomes are typically only carriers rather than affected by these X-linked recessive conditions.
Cognitive and developmental
Some degree of language learning or reading impairment may be present, and neuropsychological testing often reveals deficits in executive functions, although these deficits can often be overcome through early intervention. Also, delays in motor development may occur, which can be addressed through occupational and physical therapies. XXY males may sit up, crawl, and walk later than other infants; they may also struggle in school, both academically and with sports. It is estimated that 10% of those with Klinefelter syndrome are autistic.
Additional abnormalities may include impaired attention, reduced organizational and planning abilities, deficiencies in judgment (often presented as a tendency to interpret non-threatening stimuli as threatening), and dysfunctional decision processing.
Compared to individuals with a normal number of chromosomes, males affected by Klinefelter's syndrome may display behavioral abnormalities. These are phenotypically displayed as higher level of anxiety and depression, and mood dysregulation. These neurocognitive abnormalities are most likely due to the presence of the extra X chromosome, as indicated by studies carried out on animal models carrying an extra X chromosome.
Cause
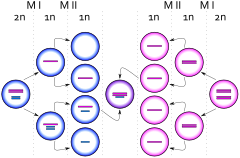

Klinefelter syndrome is not an inherited condition. Maternal age is the only known risk factor. Women at 40 years have a four times higher risk for a child with Klinefelter syndrome than women aged 24 years.
The extra chromosome is retained because of a nondisjunction event during paternal meiosis I, maternal meiosis I, or maternal meiosis II (gametogenesis). The relevant nondisjunction in meiosis I occurs when homologous chromosomes, in this case the X and Y or two X sex chromosomes, fail to separate, producing a sperm with an X and a Y chromosome or an egg with two X chromosomes. Fertilizing a normal (X) egg with this sperm produces an XXY offspring (Klinefelter). Fertilizing a double X egg with a normal sperm also produces an XXY offspring (Klinefelter).
Another mechanism for retaining the extra chromosome is through a nondisjunction event during meiosis II in the egg. Nondisjunction occurs when sister chromatids on the sex chromosome, in this case an X and an X, fail to separate. An XX egg is produced, which when fertilized with a Y sperm, yields an XXY offspring. This XXY chromosome arrangement is one of the most common genetic variations from the XY karyotype, occurring in approximately one in 500 live male births. See also Triple X syndrome.
In mammals with more than one X chromosome, the genes on all but one X chromosome are not expressed; this is known as X inactivation. This happens in XXY males, as well as normal XX females. However, in XXY males, a few genes located in the pseudoautosomal regions of their X chromosomes have corresponding genes on their Y chromosome and are capable of being expressed.
Variations
The condition 48,XXYY or 48,XXXY occurs in one in 18,000–50,000 male births. The incidence of 49,XXXXY is one in 85,000 to 100,000 male births. These variations are extremely rare. Additional chromosomal material can contribute to cardiac, neurological, orthopedic, and other anomalies.
Approximately 15–20% of males with KS may have a mosaic 47,XXY/46,XY constitutional karyotype and varying degrees of spermatogenic failure. Often, symptoms are milder in mosaic cases, with regular male secondary sex characteristics and testicular volume even falling within typical adult ranges. Another possible mosaicism is 47,XXY/46,XX with clinical features suggestive of KS and male phenotype, but this is very rare. Thus far, only approximately 10 cases of 47,XXY/46,XX have been described in literature.
Random X-inactivation
Women typically have two X chromosomes, with half of their X chromosomes switching off early in embryonic development. The same happens with people with Klinefelter's, including in both cases a small proportion of individuals with a skewed ratio between the two Xs.
Diagnosis
The standard diagnostic method is the analysis of the chromosomes' karyotype on lymphocytes. A small blood sample is sufficient as test material. In the past, the observation of the Barr body was common practice, as well. To investigate the presence of a possible mosaicism, analysis of the karyotype using cells from the oral mucosa is performed. Physical characteristics of a Klinefelter syndrome can be tall stature, low body hair, and occasionally an enlargement of the breast. Usually, a small testicle volume of 1–5 ml per testicle (standard values: 12–30 ml) occurs. During puberty and adulthood, low testosterone levels with increased levels of the pituitary hormones FSH and LH in the blood can indicate the presence of Klinefelter syndrome. A spermiogram can also be part of the further investigation. Often, an azoospermia is present, or rarely an oligospermia. Furthermore, Klinefelter syndrome can be diagnosed as a coincidental prenatal finding in the context of invasive prenatal diagnosis (amniocentesis, chorionic villus sampling). Approximately 10% of KS cases are found by prenatal diagnosis.
The symptoms of KS are often variable, so a karyotype analysis should be ordered when small testes, infertility, gynecomastia, long arms/legs, developmental delay, speech/language deficits, learning disabilities/academic issues, and/or behavioral issues are present in an individual.
Treatment
As the genetic variation is irreversible, no causal therapy is available. From the onset of puberty, the existing testosterone deficiency can be compensated by appropriate hormone-replacement therapy. Testosterone preparations are available in the form of syringes, patches, or gel. If gynecomastia is present, the surgical removal of the breast may be considered for both the psychological reasons and to reduce the risk of breast cancer.
The use of behavioral therapy can mitigate any language disorders, difficulties at school, and socialization. An approach by occupational therapy is useful in children, especially those who have dyspraxia.
Infertility treatment

Methods of reproductive medicine, such as intracytoplasmic sperm injection (ICSI) with previously conducted testicular sperm extraction (TESE), have led to men with Klinefelter syndrome producing biological offspring. By 2010, over 100 successful pregnancies have been reported using IVF technology with surgically removed sperm material from males with KS.
Prognosis
The lifespan of individuals with Klinefelter syndrome appears to be reduced by around 2.1 years compared to the general male population. These results are still questioned data, are not absolute, and need further testing.
Epidemiology
This syndrome, evenly distributed in all ethnic groups, has a prevalence of approximately four subjects per every 10,000 (0.04%) males in the general population. However, it is estimated that only 25% of the individuals with Klinefelter syndrome are diagnosed throughout their lives. The rate of Klinefelter syndrome among infertile males is 3.1%. The syndrome is also the main cause of male hypogonadism.
History
The syndrome was named after American endocrinologist Harry Klinefelter, who in 1942 worked with Fuller Albright and E. C. Reifenstein at Massachusetts General Hospital in Boston, Massachusetts, and first described it in the same year. The account given by Klinefelter came to be known as Klinefelter syndrome as his name appeared first on the published paper, and seminiferous tubule dysgenesis was no longer used. Considering the names of all three researchers, it is sometimes also called Klinefelter–Reifenstein–Albright syndrome. In 1956, Klinefelter syndrome was found to result from an extra chromosome. Plunkett and Barr found the sex chromatin body in cell nuclei of the body. This was further clarified as XXY in 1959 by Patricia Jacobs and John Anderson Strong. The first published report of a man with a 47,XXY karyotype was by Patricia Jacobs and John Strong at Western General Hospital in Edinburgh, Scotland, in 1959. This karyotype was found in a 24-year-old man who had signs of KS. Jacobs described her discovery of this first reported human or mammalian chromosome aneuploidy in her 1981 William Allan Memorial Award address.
In August 2022, a team of scientists published a study of a skeleton found at Torre Velha, Municipality of Bragança in north-eastern Portugal. The male was buried in a cut grave around 1,000CE, and found from DNA tests to be the earliest known person with this syndrome.
See also
Further reading
- Virginia Isaacs Cover (2012). Living with Klinefelter Syndrome, Trisomy X and 47,XYY: A Guide for Families and Individuals Affected by Extra X and Y Chromosomes. ISBN 978-0-615-57400-4.