
| Langerhans cell histiocytosis | |
|---|---|
 | |
| Micrograph showing a Langerhans cell histiocytosis with the characteristic reniform Langerhans cells accompanied by abundant eosinophils. H&E stain. | |
| Specialty | Hematology |
Langerhans cell histiocytosis (LCH) is an abnormal clonal proliferation of Langerhans cells, abnormal cells deriving from bone marrow and capable of migrating from skin to lymph nodes.
Symptoms range from isolated bone lesions to multisystem disease. LCH is part of a group of syndromes called histiocytoses, which are characterized by an abnormal proliferation of histiocytes (an archaic term for activated dendritic cells and macrophages). These diseases are related to other forms of abnormal proliferation of white blood cells, such as leukemias and lymphomas.
The disease has gone by several names, including Hand–Schüller–Christian disease, Abt-Letterer-Siwe disease, Hashimoto-Pritzker disease (a very rare self-limiting variant seen at birth) and histiocytosis X, until it was renamed in 1985 by the Histiocyte Society.
Classification
| Alternative names |
|---|
| Histiocytosis X
Histiocytosis X syndrome |
| Subordinate terms |
| Hand-Schüller-Christian disease
Letterer-Siwe disease |
The disease spectrum results from clonal accumulation and proliferation of cells resembling the epidermal dendritic cells called Langerhans cells, sometimes called dendritic cell histiocytosis. These cells in combination with lymphocytes, eosinophils, and normal histiocytes form typical LCH lesions that can be found in almost any organ. A similar set of diseases has been described in canine histiocytic diseases.
LCH is clinically divided into three groups: unifocal, multifocal unisystem, and multifocal multisystem.
Unifocal
Unifocal LCH, also called eosinophilic granuloma (an older term which is now known to be a misnomer), is a disease characterized by an expanding proliferation of Langerhans cells in one organ, where they cause damage called lesions. It typically has no extraskeletal involvement, but rarely a lesion can be found in the skin, lungs, or stomach. It can appear as a single lesion in an organ, up to a large quantity of lesions in one organ. When multiple lesions are scattered throughout an organ, it can be called a multifocal unisystem variety. When found in the lungs, it should be distinguished from Pulmonary Langerhans cell hystiocytosis—a special category of disease most commonly seen in adult smokers. When found in the skin it is called cutaneous single system Langerhans cell LCH. This version can heal without therapy in some rare cases. This primary bone involvement helps to differentiate eosinophilic granuloma from other forms of Langerhans Cell Histiocytosis (Letterer-Siwe or Hand-Schüller-Christian variant).
Multifocal unisystem
Seen mostly in children, multifocal unisystem LCH is characterized by fever, bone lesions and diffuse eruptions, usually on the scalp and in the ear canals. 50% of cases involve the pituitary stalk, often leading to diabetes insipidus. The triad of diabetes insipidus, exophthalmos, and lytic bone lesions is known as the Hand-Schüller-Christian triad. Peak onset is 2–10 years of age.
Multifocal multisystem
Multifocal multisystem LCH, also called Letterer-Siwe disease, is an often rapidly progressing disease in which Langerhans Cell cells proliferate in many tissues. It is mostly seen in children under age 2, and the prognosis is poor: even with aggressive chemotherapy, the five-year survival is only 50%.
Pulmonary Langerhans cell histiocytosis (PLCH)
Pulmonary Langerhans cell histiocytosis (PLCH) is a unique form of LCH in that it occurs almost exclusively in cigarette smokers. It is now considered a form of smoking-related interstitial lung disease. PLCH develops when an abundance of monoclonal CD1a-positive Langerhans (immature histiocytes) proliferate the bronchioles and alveolar interstitium, and this flood of histiocytes recruits granulocytes like eosinophils and neutrophils and agranulocytes like lymphocytes further destroying bronchioles and the interstitial alveolar space that can cause damage to the lungs. It is hypothesized that bronchiolar destruction in PLCH is first attributed to the special state of Langerhans cells that induce cytotoxic T-cell responses, and this is further supported by research that has shown an abundance of T-cells in early PLCH lesions that are CD4+ and present early activation markers. Some affected people recover completely after they stop smoking, but others develop long-term complications such as pulmonary fibrosis and pulmonary hypertension.
Signs and symptoms


LCH provokes a non-specific inflammatory response, which includes fever, lethargy, and weight loss. Organ involvement can also cause more specific symptoms.
- Bone: The most-frequently seen symptom in both unifocal and multifocal disease is painful bone swelling. The skull is most frequently affected, followed by the long bones of the upper extremities and flat bones. Infiltration in hands and feet is unusual. Osteolytic lesions can lead to pathological fractures.
- Skin: Commonly seen are a rash which varies from scaly erythematous lesions to red papules pronounced in intertriginous areas. Up to 80% of LCH patients have extensive eruptions on the scalp.
- Bone marrow: Pancytopenia with superadded infection usually implies a poor prognosis. Anemia can be due to a number of factors and does not necessarily imply bone marrow infiltration.
- Lymph node: Enlargement of the liver in 20%, spleen in 30% and lymph nodes in 50% of Histiocytosis cases.
- Endocrine glands: Hypothalamic pituitary axis commonly involved.Diabetes insipidus is most common.Anterior pituitary hormone deficiency is usually permanent.
- Lungs: some patients are asymptomatic, diagnosed incidentally because of lung nodules on radiographs; others experience chronic cough and shortness of breath.
- Less frequently gastrointestinal tract, central nervous system, and oral cavity.
Pathophysiology
The pathogenesis of Langerhans cell histiocytosis (LCH) is a matter of debate. There are ongoing investigations to determine whether LCH is a reactive (non-cancerous) or neoplastic (cancerous) process. Arguments supporting the reactive nature of LCH include the occurrence of spontaneous remissions, the extensive secretion of multiple cytokines by dendritic cells and bystander-cells (a phenomenon known as cytokine storm) in the lesional tissue, favorable prognosis and relatively good survival rate in patients without organ dysfunction or risk organ involvement.
On the other hand, the infiltration of organs by monoclonal population of pathologic cells, and the successful treatment of subset of disseminated disease using chemotherapeutic regimens are all consistent with a neoplastic process. In addition, a demonstration, using X chromosome–linked DNA probes, of LCH as a monoclonal proliferation provided additional support for the neoplastic origin of this disease. While clonality is an important attribute of cancer, its presence does not prove that a proliferative process is neoplastic. Recurrent cytogenetic or genomic abnormalities would also be required to demonstrate convincingly that LCH is a malignancy.
An activating somatic mutation of a proto-oncogene in the Raf family, the BRAF gene, was detected in 35 of 61 (57%) LCH biopsy samples with mutations being more common in patients younger than 10 years (76%) than in patients aged 10 years and older (44%). This study documented the first recurrent mutation in LCH samples. Two independent studies have confirmed this finding. Presence of this activating mutation could support the notion to characterize LCH as myeloproliferative disorder.
Diagnosis
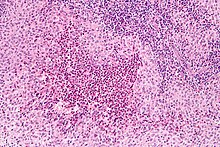
Diagnosis is confirmed histologically by tissue biopsy. Hematoxylin-eosin stain of biopsy slide will show features of Langerhans Cell e.g. distinct cell margin, pink granular cytoplasm. Presence of Birbeck granules on electron microscopy and immuno-cytochemical features e. g. CD1 positivity are more specific. Initially routine blood tests e.g. full blood count, liver function test, U&Es, bone profile are done to determine disease extent and rule out other causes.
Imaging may be evident in chest X-rays with micronodular and reticular changes of the lungs with cyst formation in advanced cases. MRI and High-resolution CT may show small, cavitated nodules with thin-walled cysts. MRI scan of the brain can show three groups of lesions such as tumourous/granulomatous lesions, nontumourous/granulomatous lesions, and atrophy. Tumourous lesions are usually found in the hypothalamic-pituitary axis with space-occupying lesions with or without calcifications. In non-tumourous lesions, there is a symmetrical hyperintense T2 signal with hypointense or hyperintense T1 signal extending from grey matter into the white matter. In the basal ganglia, MRI shows a hyperintense T1 signal in the globus pallidus.
Assessment of endocrine function and bonemarrow biopsy are also performed when indicated.
- S-100 protein is expressed in a cytoplasmic pattern
- peanut agglutinin (PNA) is expressed on the cell surface and perinuclearly
- major histocompatibility (MHC) class II is expressed (because histiocytes are macrophages)
- CD1a
- langerin (CD207), a Langerhans Cell–restricted protein that induces the formation of Birbeck granules and is constitutively associated with them, is a highly specific marker.
Treatment
Guidelines for management of patients up to 18 years with Langerhans cell histiocytosis have been suggested. Treatment is guided by extent of disease. Solitary bone lesion may be amenable through excision or limited radiation, dosage of 5-10 Gy for children, 24-30 Gy for adults. However systemic diseases often require chemotherapy. Use of systemic steroid is common, singly or adjunct to chemotherapy. Local steroid cream is applied to skin lesions. Endocrine deficiency often require lifelong supplement e.g. desmopressin for diabetes insipidus which can be applied as nasal drop. Chemotherapeutic agents such as alkylating agents, antimetabolites, vinca alkaloids either singly or in combination can lead to complete remission in diffuse disease.
Prognosis
There is a general excellent prognosis for the disease as those with localized disease have a long life span. With multi-focal disease 60% have a chronic course, 30% achieve remission and mortality is up to 10%. A full recovery can be expected for people who seek treatment and do not have more lesions at 12 and 24 months. However, 50% of children under 2 with disseminated Langerhans cell histiocytosis die of the disease. The prognosis rate decreases for patients who experience lung involvement. Whereas patients with skin and a solitary lymph node involvement generally have a good prognosis. Although there is a general good prognosis for Langerhans cell histiocytosis, approximately 0% of patients with the disease are prone to various complications such as musculoskeletal disability, skin scarring and diabetes insipidus.
Prevalence
LCH usually affects children between 1 and 15 years old, with a peak incidence between 5 and 10 years of age. Among children under the age of 10, yearly incidence is thought to be 1 in 200,000; and in adults even rarer, in about 1 in 560,000. It has been reported in elderly but is vanishingly rare. It is most prevalent in Caucasians, and affects males twice as often as females. In other populations too the prevalence in males is slightly more than in females.
LCH is usually a sporadic and non-hereditary condition but familial clustering has been noted in limited number of cases. Hashimoto-Pritzker disease is a congenital self-healing variant of Hand-Schüller-Christian disease.
Culture
In the 10th episode of season 3 of House entitled "Merry Little Christmas", the primary patient is a girl with dwarfism who has a variety of symptoms, who is ultimately diagnosed with Langerhans cell histiocytosis. Also in the 5th episode, season 1 of "The Good Doctor", Dr. Murphy tries to diagnose Langerhans cell histiocytosis in a boy with a previously diagnosed osteosarcoma.In an episode of Mystery Diagnosis, "The Woman Who Saw Pink", Brooke Rohrer has experiencing symptoms of abdominal pain, was diagnosed with Langerhans cell histiocytosis.
Nomenclature
Langerhans cell histiocytosis is occasionally misspelled as "Langerhan" or "Langerhan's" cell histiocytosis, even in authoritative textbooks. The name, however, originates back to its discoverer, Paul Langerhans.