
| MERRF syndrome | |
|---|---|
| Other names | Fukuhara syndrome |
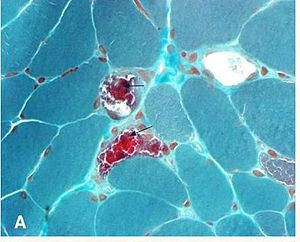 | |
| "ragged red fibers" in MERRF syndrome | |
| Specialty |
Neurology |
MERRF syndrome (or myoclonic epilepsy with ragged red fibers) is a mitochondrial disease. It is extremely rare, and has varying degrees of expressivity owing to heteroplasmy. MERRF syndrome affects different parts of the body, particularly the muscles and nervous system. The signs and symptoms of this disorder appear at an early age, generally childhood or adolescence. The causes of MERRF syndrome are difficult to determine, but because it is a mitochondrial disorder, it can be caused by the mutation of nuclear DNA or mitochondrial DNA. The classification of this disease varies from patient to patient, since many individuals do not fall into one specific disease category. The primary features displayed on a person with MERRF include myoclonus, seizures, cerebellar ataxia, myopathy, and ragged red fibers (RRF) on muscle biopsy, leading to the disease's name. Secondary features include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, or multiple lipomata. Mitochondrial disorders, including MERRFS, may present at any age.
Symptoms and signs
An individual displaying MERRFs syndrome will manifest not only a single symptom, but patients regularly display more than one affected body part at a time. It has been observed that patients with MERRF syndrome will primarily display myoclonus as a first symptom. There may also be seizures, cerebellar ataxia and myopathy. Secondary features can include dementia, optic atrophy, bilateral deafness, peripheral neuropathy, spasticity, multiple lipomata, and/or cardiomyopathy with Wolff Parkinson-White syndrome. Most patients will not exhibit all of these symptoms, but more than one of these symptoms will be present in a patient who has been diagnosed with MERRF disease. Mitochondrial disorders, including MERRF, may present at any age. Due to the multiple symptoms presented by the individual, the severity of the syndrome is very difficult to evaluate.
Causes

The cause of MERRF disorder is due to mutations in the mitochondrial genome. This means that it is a pathological variant in mtDNA (mitochondrial DNA) and is transmitted by maternal inheritance. Four point mutations in the genome can be identified that are associated with MERRF: m.A8344G, m.T8356C, m.G8361A, and m.G8363A. The point mutation m.A8344G is most commonly associated with MERRF, in a study published by Paul Jose Lorenzoni from the Department of neurology at University of Panama stated that 80% of the patients with MERRF disease exhibited this point mutation. This point mutation disrupts the mitochondrial gene for tRNA-Lys. This disrupts the synthesis of proteins. The remaining mutations only account for 10% of cases, and the remaining 10% of the patients with MERRF did not have an identifiable mutation in the mitochondrial DNA.
Many genes are involved. These genes include:
It involves the following characteristics:
- progressive myoclonic epilepsy
- "Ragged Red Fibers" - clumps of diseased mitochondria accumulate in the subsarcolemmal region of the muscle fiber and appear as "Ragged Red Fibers" when muscle is stained with modified Gömöri trichrome stain.
There is currently no cure for MERRF.
Mechanism
The mechanism by which MERRFs syndrome occur is not yet well understood. The human mitochondrial tRNA mutations are associated with a variety of diseases including mitochondrial myopathies. However, it is understood that defects in the mitochondrial DNA (mtDNA) have been associated with these diseases, and studies have been able to assign biochemical defects. One of these defects has to do with the decreased energy available for cell processes. As muscles are stained with Gömöri trichrome, characteristic ragged red fibers are visible under the microscope. This appearance is due to the accumulation of abnormal mitochondria below the plasma membrane of the muscle fiber. These may extend throughout the muscle fiber as the disease severity increases. The mitochondrial aggregates cause the contour of the muscle fiber to become irregular, leading to the "ragged" appearance.
Diagnosis
The diagnosis varies from individual to individual. Each is evaluated and diagnosed according to age, clinical phenotype, and pressed inheritance pattern. If the individual has been experiencing myoclonus, the doctor will run a series of genetic studies to determine if it is a mitochondrial disorder.
The molecular genetic studies are run to identify the reason of for the mutations underlying the mitochondrial dysfunction. This approach will avoid the need for a muscle biopsy or an exhaustive metabolic evaluation. After sequencing the mitochondrial genomes, four points mutations in the genome can be identified which are associated with MERRF: A8344G, T8356C, G8361A, and G8363A. The point mutation A8344G is mostly associated with MERRF, in a study published by Paul Jose Lorenzoni from the Department of neurology at University of Panama stated that 80% of the patients with MERRF disease exhibited this point mutation. The remaining mutations only account for 10% of cases, and the remaining 10% of the patients with MERRF did not have an identifiable mutation in the mitochondrial DNA.
If a patient does not exhibit mitochondrial DNA mutations, there are other ways that they can be diagnosed with MERRF. They can go through computed tomography (CT) or magnetic resonance imaging (MRI).The classification for the severity of MERRF syndrome is difficult to distinguish since most individuals will exhibit multi-symptoms. This is often necessary for children with complex neurologic or multi-system involvement, as described below.
History and physical examination of the patient
A detailed family history should be obtained from at least three generations, particularly if there have been any neonatal and childhood deaths. A family history may also indicate if any family members exhibit features of the multi-system disease, specifically if there has been maternal inheritance. This would show transmission of the disease only to females, or if there is a family member who experienced a multi-system involvement such as:brain condition that a family member has been record to have such as seizures, dystonia, ataxia, or stroke-like episodes. There may also be optic atrophy, skeletal muscle with a history of myalgia, weakness, or ptosis. Family history may also include neuropathy and dysautonomia, or heart conditions such as cardiomyopathy. The patient's history might also exhibit kidney problems, such as proximal nephron dysfunction. There may also be endocrine conditions, such as diabetes or hypoparathyroidism. The patient might have also had a gastrointestinal condition which could have been due to liver disease, as well as episodes of nausea or vomiting. Multiple lipomas in the skin, sideroblastic anemia and pancytopenia in the metabolic system, or short stature might all be examples of patients with possible symptoms of MERRF disease.
Treatment
Like many mitochondrial diseases, there is no cure for MERRF, no matter the means for diagnosis of the disease. The treatment is primarily symptomatic. High doses of coenzyme Q10, B complex vitamins, and L-Carnitine are used for the altered metabolic processing that results in the disease. There is very little success with these treatments as therapies in hopes of improving mitochondrial function. The treatment only alleviates symptoms, and these do not prevent the disease from progressing. Patients with concomitant disease, such as diabetes, deafness, or cardiac disease, are treated in combination to manage symptoms.
Research
The Journal of Child Neurology published a paper that discusses possible new methods to test for MERRF and other mitochondrial diseases through a simple swabbing technique. This is a less invasive technique which allows for an analysis of buccal mitochondrial DNA, and showed significant amounts of the common 5 kb and 7.4 kb mitochondrial DNA deletions, which are also detectable in blood. This study suggests that a buccal swab approach can be used to informatively examine mitochondrial dysfunction in children with seizures and may be applicable to screening mitochondrial disease with other clinical presentations.
Proceedings of the National Academy of Science of the United States of America published an article investigating the human mitochondrial tRNA (hmt-tRNA) mutations which are associated with mitochondrial myopathies. Since the current understanding of the precise molecular mechanisms of these mutations is limited, there is no efficient method to treat their associated mitochondrial diseases. All pathogenic mutants displayed pleiotropic phenotypes, with the exception of the G34A anticodon mutation, which solely affected aminoacylation.
See also
External links
- MERRF+Syndrome at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- merrf at NIH/UW GeneTests