
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Istodax |
| Other names | FK228; FR901228; Istodax |
| MedlinePlus | a610005 |
| License data |
|
| Routes of administration |
Intravenous infusion |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Not applicable (IV only) |
| Protein binding | 92–94% |
| Metabolism | Liver (mostly CYP3A4-mediated) |
| Elimination half-life | 3 hours |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.211.884 |
| Chemical and physical data | |
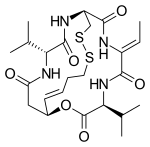
| Formula | C24H36N4O6S2 |
| Molar mass | 540.69 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
|
| |
Romidepsin, sold under the brand name Istodax, is an anticancer agent used in cutaneous T-cell lymphoma (CTCL) and other peripheral T-cell lymphomas (PTCLs). Romidepsin is a natural product obtained from the bacterium Chromobacterium violaceum, and works by blocking enzymes known as histone deacetylases, thus inducing apoptosis. It is sometimes referred to as depsipeptide, after the class of molecules to which it belongs. Romidepsin is branded and owned by Gloucester Pharmaceuticals, a part of Celgene.
History
Romidepsin was first reported in the scientific literature in 1994, by a team of researchers from Fujisawa Pharmaceutical Company (now Astellas Pharma) in Tsukuba, Japan, who isolated it in a culture of Chromobacterium violaceum from a soil sample obtained in Yamagata Prefecture. It was found to have little to no antibacterial activity, but was potently cytotoxic against several human cancer cell lines, with no effect on normal cells; studies on mice later found it to have antitumor activity in vivo as well.
The first total synthesis of romidepsin was accomplished by Harvard researchers and published in 1996. Its mechanism of action was elucidated in 1998, when researchers from Fujisawa and the University of Tokyo found it to be a histone deacetylase inhibitor with effects similar to those of trichostatin A.
Clinical trials
Phase I studies of romidepsin, initially codenamed FK228 and FR901228, began in 1997.Phase II and phase III trials were conducted for a variety of indications. The most significant results were found in the treatment of cutaneous T-cell lymphoma (CTCL) and other peripheral T-cell lymphomas (PTCLs).
In 2004, romidepsin received Fast Track designation from the FDA for the treatment of cutaneous T-cell lymphoma, and orphan drug status from the FDA and the European Medicines Agency for the same indication.
The FDA approved romidepsin for CTCL in November 2009 and approved romidepsin for other peripheral T-cell lymphomas (PTCLs) in June 2011.
A randomised, phase III trial of romidepsin + CHOP chemotherapy vs CHOP chemotherapy for patients with peripheral T cell lymphoma returned negative results, having no significant impact on progression free survival or overall survival.
Pre-clinical HIV study
In 2014, PLOS Pathogens published a study involving romidepsin in a trial designed to reactivate latent HIV virus in order to deplete the HIV reservoir. Latently infected T-cells were exposed in vitro and ex vivo to romidepsin, leading to an increase in detectable levels of cell-associated HIV RNA. The trial also compared the effect of romidepsin to another histone deacetylase inhibitor, Vorinostat
Autism study in animal model
A study involving romidepsin in an animal study that showed that a brief treatment with low amounts of romidepsin could reverse social deficits in a mouse model of autism.
Pharmacodynamics
In a Phase II trial of romidepsin involving patients with CTCL or PTCL, there was evidence of increased histone acetylation in peripheral blood mononuclear cells (PBMCs) extending 4–48 hours. Expression of the ABCB1 gene, a marker of romidepsin-induced gene expression, was also increased in both PBMCs and tumor biopsy samples. Increased gene expression following increased histone acetylation is an expected effect of an HDAC inhibitor. Increased hemoglobin F (another surrogate marker for gene-expression changes resulting from HDAC inhibition) was also detected in blood after romidepsin administration, and persistent histone acetylation was inversely associated with drug clearance and directly associated with patient response to therapy.
Dosage and administration
The approved dosage of romidepsin in both CTCL and PTCL is a four-hour i.v. administration of 14 mg/m2 on days 1, 8, and 15 of a 28-day treatment cycle. This cycle should be repeated as long as the patient continues to benefit and tolerate the therapy. A dose reduction to 10 mg/m2 is possible in some patients who experience high-grade toxicities.
Pharmacokinetics
In trials involving patients with advanced cancers, romidepsin exhibited linear pharmacokinetics across doses ranging from 1.0 to 24.9 mg/m2 when administered intravenously over four hours. Age, race, sex, mild-to-severe renal impairment, and mild-to-moderate hepatic impairment had no effect on romidepsin pharmacokinetics. No accumulation of plasma concentration was observed after repeated dosing.
Mechanism of action
Romidepsin acts as a prodrug with the disulfide bond undergoing reduction within the cell to release a zinc-binding thiol. The thiol binds to a zinc atom in the binding pocket of Zn-dependent histone deacetylase to block its activity. Thus it is an HDAC inhibitor. Many HDAC inhibitors are potential treatments for cancer through the ability to epigenetically restore normal expression of tumor suppressor genes, which may result in cell cycle arrest, differentiation, and apoptosis.
Adverse effects
The use of romidepsin is uniformly associated with adverse effects. In clinical trials, the most common were nausea and vomiting, fatigue, infection, loss of appetite, and blood disorders (including anemia, thrombocytopenia, and leukopenia). It has also been associated with infections, and with metabolic disturbances (such as abnormal electrolyte levels), skin reactions, altered taste perception, and changes in cardiac electrical conduction.