
| Uveal melanoma | |
|---|---|
| Other names | Ocular melanoma |
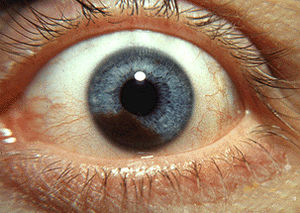 | |
| Iris melanoma | |
| Specialty | Oncology |
| Symptoms | A sensation of flashes or specks of dust (floaters); growing dark spot on the iris; change in the shape of the pupil; poor or blurry vision in one eye; loss of peripheral vision in one eye. |
| Complications | Retinal detachment |
| Usual onset | Visual abnormalities |
| Types | choroid, iris, and ciliary body |
| Diagnostic method | clinical examination by biomicroscopy and indirect ophthalmoscopy |
| Differential diagnosis | For choroid: choroidal tumors, especially choroidal nevus, metastatic tumors, choroidal hemangioma, and osteoma; hemorrhagic conditions like AMD and hemorrhagic choroidal detachment; retinal tumors such as congenital retinal pigment epithelium hypertrophy and retinal pigment epithelium adenocarcinoma; and inflammatory lesions like posterior scleritis. For iris: iris nevus, iris pigment epithelial cyst, iris stromal cyst, metastatic tumor of the iris, melanocytoma, iris atrophy and Cogan-Reese syndrome. For ciliary body: staphyloma, medulloepithelioma and leiomyoma. |
| Prevention | Reduce UV exposure to the eye. |
| Treatment | Brachytherapy, enucleation, proton beam radiotherapy, transpupillary thermotherapy, photocoagulation, photodynamic therapy, and local resection. |
| Frequency | 5 cases per million people per year |
Uveal melanoma is a type of eye cancer in the uvea of the eye. It is traditionally classed as originating in the iris, choroid, and ciliary body, but can also be divided into class I (low metastatic risk) and class II (high metastatic risk). Symptoms include blurred vision, loss of vision or photopsia, but there may be no symptoms.
Tumors arise from the pigment cells (melanocytes) that reside within the uvea and give color to the eye. These melanocytes are distinct from the retinal pigment epithelium cells underlying the retina that do not form melanomas. When eye melanoma is spread to distant parts of the body, the five-year survival rate is about 15%.
It is the most common type of primary eye cancer. Males and females are affected equally. Worldwide, there are around 2 million to 8 million cases per year. More than 50% spread, mostly to the liver.
Signs and symptoms
Ocular melanoma may present without symptoms depending upon the location and size of the tumor. When symptoms do occur, they can include:
- blurred vision
- double vision (diplopia)
- irritation
- pain
- a perception of flashes of light in the eye (photopsia)
- a reduction in the total field of vision
- loss of vision
- a sensation of a foreign body in the field of vision (floaters)
- redness, bulging or displacement of the eye (proptosis)
- a change in the shape of the pupil
- pressure within the eye
- metamorphopsia (a distortion of vision where, when a person looks at a grid of straight lines, the lines appear wavy and parts of the grid appears blank).
Types
Uveal melanomas, often referred to by the media and in the general population as ocular melanomas, may arise from any of the three parts of the uvea, and are sometimes referred to by their location, choroidal melanoma, ciliary body melanoma, or iris melanoma. Large tumors often encompass multiple parts of the uvea and can be named accordingly. True iris melanomas, originating from within the iris as opposed to originating elsewhere and invading the iris, are distinct in their etiology and prognosis, such that the other tumors are often referred to collectively as posterior uveal melanomas.
Iris melanoma
Uveal tumors can originate from melanocytes residing within the iris. Benign melanocytic tumors, such as iris freckles and moles (nevi), are common and pose no health risks, unless they show signs of malignancy, in which case they are classified as iris melanomas. Though derived from uveal melanocytes, iris melanomas share more in common with cutaneous (skin) melanomas in that they frequently harbor BRAF mutations associated with ultraviolet damage. Iris melanomas are much less likely to metastasize than other uveal melanomas, and less likely to impair vision if detected and treated early. Approximately 5% of uveal melanomas involve the iris.
Posterior uveal melanoma

Benign melanocytic tumors of the choroid, such as choroidal freckles and nevi, are very common and pose no health risks, unless they show signs of malignancy, in which case they are considered melanomas. Uveal melanoma is distinct from most skin melanomas associated with ultraviolet exposure; however, it shares several similarities with non-sun-exposed melanomas, such as acral melanomas and mucosal melanomas. BRAF mutations are extremely rare in posterior uveal melanomas; instead, uveal melanomas frequently harbor GNAQ/GNA11 mutations, a trait shared with blue nevi, nevus of Ota, and ocular melanosis. As seen in BRAF, mutations in GNAQ/GNA11 are early events in tumorigenesis and are not prognostic for tumor stage or later metastatic spread. In contrast, mutations in the gene BAP1 are strongly linked to metastatic spread and patient survival. Incidence of posterior uveal melanoma is highest among people with light skin and blue eyes. Other risk factors, such as blue light exposure and arc welding, have been put forward, but are still debated in the field. Mobile phone use is not a risk factor for uveal melanoma.

Cause
The cause of uveal melanoma is unclear. Uveal nevi are common (5% of Caucasians), but rarely progress to melanoma.
Metastasis
Because there are no lymphatic channels to the uveal tract, metastasis occurs through local extension and/or blood-borne dissemination. The most common site of metastasis for uveal melanoma is the liver; the liver is the first site of metastasis for 80%-90% of ocular melanoma patients. Other common sites of metastasis include the lung, bones, and just beneath the skin (subcutaneous). Approximately 50 percent of patients will develop metastases within 15 years after treatment of the primary tumor, and the liver will be involved 90% of the time. Metastasis can occur more than 10 years after treatment of the primary tumor, and patients should not be considered cured even after a 10-year interval of monitoring. Molecular features of the tumor, including chromosome 3 status, chromosome 6p status, and chromosome 8q status and gene expression profiling (such as the DecisionDx-UM test), can be used to adjust this likelihood of metastasis for an individual patient.
The average survival time after diagnosis of liver metastases depends on the extent of systemic spread. The disease-free interval, the performance status, the liver substitution by metastases and the serum level of lactic dehydrogenase are the most important prognostic factors for metastatic uveal melanoma. There is currently no cure for metastatic uveal melanoma.
Treatment
The treatment protocol for uveal melanoma has been directed by many clinical studies, the most important being The Collaborative Ocular Melanoma Study (COMS). The treatment varies depending upon many factors, chief among them the size of the tumor and results from testing of biopsied material from the tumor. Primary treatment can involve removal of the affected eye (enucleation); however, this is now reserved for cases of extreme tumor burden or other secondary problems. Advances in radiation therapies have significantly decreased the number of patients treated by enucleation in developed countries.
Plaque brachytherapy
The most common radiation treatment is plaque brachytherapy, in which a small disc-shaped shield (plaque) encasing radioactive seeds (most often iodine-125, though ruthenium-106 and palladium-103 are also used) is attached to the outside surface of the eye, overlying the tumor. The plaque is left in place for a few days and then removed. The risk of metastasis after plaque radiotherapy is the same as that of enucleation, suggesting that micrometastatic spread occurs prior to treatment of the primary tumor.
Proton therapy
Proton therapy delivers powerful doses of radiation to the tumor while sparing surrounding healthy eye tissue. The treatment is also effective at controlling tumor growth and maintaining a patient's natural eye.
Immunotherapy
On January 25, 2022, the FDA approved tebentafusp (Kimmtrak) for adult HLA-A*2-positive patients with metastatic uveal melanoma. Approval was based on the IMCgp100-202 trial (NCT03070392), which compared tebentafusp with investigator's choice of either dacarbazine, pembrolizumab or ipilimumab. Patients lived a median of 16 months when treated with investigator's choice of therapy, compared to a median 21.7 months with tebentafusp. This translated in a 49% reduction in the risk of death. Overall survival after one year of treatment was 59% in the control group, versus 73% in the tebentafusp group.
Other
Other modalities of treatment include transpupillary thermotherapy, resection of the tumor, gamma knife stereotactic radiosurgery, or a combination of different modalities. Different surgical resection techniques can include trans-scleral partial choroidectomy and transretinal endoresection.
A 2017 analysis of genomic data led to a new analysis of clinical subtyping in uveal melanoma. Ocular melanoma expert Professor Sarah Coupland, in 2018, suggested cautious optimism as new types of targeted therapeutics are tested and approved.
Prognosis
When eye melanoma is spread to distant parts of the body, the five-year survival rate is about 15%.
Several clinical and pathological prognostic factors have been identified that are associated with higher risk of metastasis of uveal melanomas. These include large tumor size, ciliary body involvement, presence of orange pigment overlying the tumor, and older patient age. Likewise several histological and cytological factors are associated with higher risk of metastasis, including presence and extent of cells with epithelioid morphology, presence of looping extracellular matrix patterns, increased infiltration of immune cells, and staining with several immunohistochemical markers.
The most important genetic alteration associated with poor prognosis in uveal melanoma is inactivation of BAP1, which most often occurs through mutation of one allele and subsequent loss of an entire copy of chromosome 3 (monosomy 3) to unmask the mutant copy. Because of this function in inactivation of BAP1, monosomy 3 correlates strongly with metastatic spread Where BAP1 mutation status is not available, gains on chromosomes 6 and 8 can be used to refine the predictive value of the monosomy 3 screen, with gain of 6p indicating a better prognosis and gain of 8q indicating a worse prognosis in disomy 3 tumors. In rare instances, monosomy 3 tumors may duplicate the BAP1-mutant copy of the chromosome to return to a disomic state referred to as isodisomy. Thus, isodisomy 3 is prognostically equivalent to monosomy 3, and both can be detected by tests for chromosome 3 loss of heterozygosity. Monosomy 3, along with other chromosomal gains, losses, amplifications, and LOH, can be detected in fresh or paraffin-embedded samples by virtual karyotyping.
The most accurate prognostic factor is molecular classification by gene expression profiling of uveal melanomas. This analysis has been used to identify two subclasses of uveal melanomas: class 1 tumors that have a very low risk of metastasis, and class 2 tumors that have a very high risk of metastasis. Gene expression profiling outperforms all of the above-mentioned factors at predicting metastatic spread of the primary tumor, including monosomy 3.
Surveillance
Currently, there is no consensus regarding type or frequency of scans following diagnosis and treatment of the primary eye tumor.
Of the 50% of patients who develop metastatic disease, more than 90% of patients will develop liver metastases. As such, the majority of surveillance techniques are focused on the liver. These include abdominal magnetic resonance imaging (MRI), abdominal ultrasound and liver function tests. The scientific community is currently working to develop guidelines, but until then, each patient must take into consideration their individual clinical situation and discuss appropriate surveillance with their doctors.
Some ophthalmologists have also found promise with the use of intravitreal avastin injections in patients with radiation-induced retinopathy, a side effect of plaque brachytherapy treatment, as well as imaging surveillance with SD-OCT.
Epidemiology
Uveal melanomas are the most common primary intraocular tumor in adults. Uveal melanoma is classified as a rare cancer with 5.1 cases per million people per year. The incidence has remained stable for several years.
There are about 2500 patients with UM diagnosed annually in the US.
Apparent clustering
In 2018, it was reported that two clusters of cases have been identified in Huntersville, North Carolina, and Auburn, Alabama. Cases involved alumni of Auburn University who were acquainted with each other. Health officials investigated and found that the "cluster" is an artifact of social networking.
History
Uveal melanoma was first described in the literature in 1809-1812 by two Scottish surgeons, Allan Burns and James Wardrop.