

Artificial gene synthesis, or simply gene synthesis, refers to a group of methods that are used in synthetic biology to construct and assemble genes from nucleotides de novo. Unlike DNA synthesis in living cells, artificial gene synthesis does not require template DNA, allowing virtually any DNA sequence to be synthesized in the laboratory. It comprises two main steps, the first of which is solid-phase DNA synthesis, sometimes known as DNA printing. This produces oligonucleotide fragments that are generally under 200 base pairs. The second step then involves connecting these oligonucleotide fragments using various DNA assembly methods. Because artificial gene synthesis does not require template DNA, it is theoretically possible to make a completely synthetic DNA molecule with no limits on the nucleotide sequence or size.
Synthesis of the first complete gene, a yeast tRNA, was demonstrated by Har Gobind Khorana and coworkers in 1972. Synthesis of the first peptide- and protein-coding genes was performed in the laboratories of Herbert Boyer and Alexander Markham, respectively. More recently, artificial gene synthesis methods have been developed that will allow the assembly of entire chromosomes and genomes. The first synthetic yeast chromosome was synthesised in 2014, and entire functional bacterial chromosomes have also been synthesised. In addition, artificial gene synthesis could in the future make use of novel nucleobase pairs (unnatural base pairs).
Standard methods for DNA synthesis
Oligonucleotide synthesis
Oligonucleotides are chemically synthesized using building blocks called nucleoside phosphoramidites. These can be normal or modified nucleosides which have protecting groups to prevent their amines, hydroxyl groups and phosphate groups from interacting incorrectly. One phosphoramidite is added at a time, the 5' hydroxyl group is deprotected and a new base is added and so on. The chain grows in the 3' to 5' direction, which is backwards relative to biosynthesis. At the end, all the protecting groups are removed. Nevertheless, being a chemical process, several incorrect interactions occur leading to some defective products. The longer the oligonucleotide sequence that is being synthesized, the more defects there are, thus this process is only practical for producing short sequences of nucleotides. The current practical limit is about 200 bp (base pairs) for an oligonucleotide with sufficient quality to be used directly for a biological application. HPLC can be used to isolate products with the proper sequence. Meanwhile, a large number of oligos can be synthesized in parallel on gene chips. For optimal performance in subsequent gene synthesis procedures they should be prepared individually and in larger scales.
Annealing based connection of oligonucleotides
Usually, a set of individually designed oligonucleotides is made on automated solid-phase synthesizers, purified and then connected by specific annealing and standard ligation or polymerase reactions. To improve specificity of oligonucleotide annealing, the synthesis step relies on a set of thermostable DNA ligase and polymerase enzymes. To date, several methods for gene synthesis have been described, such as the ligation of phosphorylated overlapping oligonucleotides, the Fok I method and a modified form of ligase chain reaction for gene synthesis. Additionally, several PCR assembly approaches have been described. They usually employ oligonucleotides of 40-50 nucleotides length that overlap each other. These oligonucleotides are designed to cover most of the sequence of both strands, and the full-length molecule is generated progressively by overlap extension (OE) PCR, thermodynamically balanced inside-out (TBIO) PCR or combined approaches. The most commonly synthesized genes range in size from 600 to 1,200 bp although much longer genes have been made by connecting previously assembled fragments of under 1,000 bp. In this size range it is necessary to test several candidate clones confirming the sequence of the cloned synthetic gene by automated sequencing methods.
Limitations
Moreover, because the assembly of the full-length gene product relies on the efficient and specific alignment of long single stranded oligonucleotides, critical parameters for synthesis success include extended sequence regions comprising secondary structures caused by inverted repeats, extraordinary high or low GC-content, or repetitive structures. Usually these segments of a particular gene can only be synthesized by splitting the procedure into several consecutive steps and a final assembly of shorter sub-sequences, which in turn leads to a significant increase in time and labor needed for its production. The result of a gene synthesis experiment depends strongly on the quality of the oligonucleotides used. For these annealing based gene synthesis protocols, the quality of the product is directly and exponentially dependent on the correctness of the employed oligonucleotides. Alternatively, after performing gene synthesis with oligos of lower quality, more effort must be made in downstream quality assurance during clone analysis, which is usually done by time-consuming standard cloning and sequencing procedures. Another problem associated with all current gene synthesis methods is the high frequency of sequence errors because of the usage of chemically synthesized oligonucleotides. The error frequency increases with longer oligonucleotides, and as a consequence the percentage of correct product decreases dramatically as more oligonucleotides are used. The mutation problem could be solved by shorter oligonucleotides used to assemble the gene. However, all annealing based assembly methods require the primers to be mixed together in one tube. In this case, shorter overlaps do not always allow precise and specific annealing of complementary primers, resulting in the inhibition of full length product formation. Manual design of oligonucleotides is a laborious procedure and does not guarantee the successful synthesis of the desired gene. For optimal performance of almost all annealing based methods, the melting temperatures of the overlapping regions are supposed to be similar for all oligonucleotides. The necessary primer optimisation should be performed using specialized oligonucleotide design programs. Several solutions for automated primer design for gene synthesis have been presented so far.
Error correction procedures
To overcome problems associated with oligonucleotide quality several elaborate strategies have been developed, employing either separately prepared fishing oligonucleotides, mismatch binding enzymes of the mutS family or specific endonucleases from bacteria or phages. Nevertheless, all these strategies increase time and costs for gene synthesis based on the annealing of chemically synthesized oligonucleotides.
Massively parallel sequencing has also been used as a tool to screen complex oligonucleotide libraries and enable the retrieval of accurate molecules. In one approach, oligonucleotides are sequenced on the 454 pyrosequencing platform and a robotic system images and picks individual beads corresponding to accurate sequence. In another approach, a complex oligonucleotide library is modified with unique flanking tags before massively parallel sequencing. Tag-directed primers then enable the retrieval of molecules with desired sequences by dial-out PCR.
Increasingly, genes are ordered in sets including functionally related genes or multiple sequence variants on a single gene. Virtually all of the therapeutic proteins in development, such as monoclonal antibodies, are optimised by testing many gene variants for improved function or expression.
Unnatural base pairs
While traditional nucleic acid synthesis only uses 4 base pairs - adenine, thymine, guanine and cytosine, oligonucleotide synthesis in the future could incorporate the use of unnatural base pairs, which are artificially designed and synthesized nucleobases that do not occur in nature.
In 2012, a group of American scientists led by Floyd Romesberg, a chemical biologist at the Scripps Research Institute in San Diego, California, published that his team designed an unnatural base pair (UBP). The two new artificial nucleotides or Unnatural Base Pair (UBP) were named d5SICS and dNaM. More technically, these artificial nucleotides bearing hydrophobic nucleobases, feature two fused aromatic rings that form a (d5SICS–dNaM) complex or base pair in DNA. In 2014 the same team from the Scripps Research Institute reported that they synthesized a stretch of circular DNA known as a plasmid containing natural T-A and C-G base pairs along with the best-performing UBP Romesberg's laboratory had designed, and inserted it into cells of the common bacterium E. coli that successfully replicated the unnatural base pairs through multiple generations. This is the first known example of a living organism passing along an expanded genetic code to subsequent generations. This was in part achieved by the addition of a supportive algal gene that expresses a nucleotide triphosphate transporter which efficiently imports the triphosphates of both d5SICSTP and dNaMTP into E. coli bacteria. Then, the natural bacterial replication pathways use them to accurately replicate the plasmid containing d5SICS–dNaM.
The successful incorporation of a third base pair is a significant breakthrough toward the goal of greatly expanding the number of amino acids which can be encoded by DNA, from the existing 20 amino acids to a theoretically possible 172, thereby expanding the potential for living organisms to produce novel proteins. In the future, these unnatural base pairs could be synthesised and incorporated into oligonucleotides via DNA printing methods.
DNA assembly
DNA printing can thus be used to produce DNA parts, which are defined as sequences of DNA that encode a specific biological function (for example, promoters, transcription regulatory sequences or open reading frames). However, because oligonucleotide synthesis typically cannot accurately produce oligonucleotides sequences longer than a few hundred base pairs, DNA assembly methods have to be employed to assemble these parts together to create functional genes, multi-gene circuits or even entire synthetic chromosomes or genomes. Some DNA assembly techniques only define protocols for joining DNA parts, while other techniques also define the rules for the format of DNA parts that are compatible with them. These processes can be scaled up to enable the assembly of entire chromosomes or genomes. In recent years, there has been proliferation in the number of different DNA assembly standards with 14 different assembly standards developed as of 2015, each with their pros and cons. Overall, the development of DNA assembly standards has greatly facilitated the workflow of synthetic biology, aided the exchange of material between research groups and also allowed for the creation of modular and reusable DNA parts.
The various DNA assembly methods can be classified into three main categories – endonuclease-mediated assembly, site-specific recombination, and long-overlap-based assembly. Each group of methods has its distinct characteristics and their own advantages and limitations.
Endonuclease-mediated assembly
Endonucleases are enzymes that recognise and cleave nucleic acid segments and they can be used to direct DNA assembly. Of the different types of restriction enzymes, the type II restriction enzymes are the most commonly available and used because their cleavage sites are located near or in their recognition sites. Hence, endonuclease-mediated assembly methods make use of this property to define DNA parts and assembly protocols.
BioBricks

The BioBricks assembly standard was described and introduced by Tom Knight in 2003 and it has been constantly updated since then. Currently, the most commonly used BioBricks standard is the assembly standard 10, or BBF RFC 10. BioBricks defines the prefix and suffix sequences required for a DNA part to be compatible with the BioBricks assembly method, allowing the joining of all DNA parts which are in the BioBricks format.
The prefix contains the restriction sites for EcoRI, NotI and XBaI, while the suffix contains the SpeI, NotI and PstI restriction sites. Outside of the prefix and suffix regions, the DNA part must not contain these restriction sites. To join two BioBrick parts together, one of the plasmids is digested with EcoRI and SpeI while the second plasmid is digested with EcoRI and XbaI. The two EcoRI overhangs are complementary and will thus anneal together, while SpeI and XbaI also produce complementary overhangs which can also be ligated together. As the resulting plasmid contains the original prefix and suffix sequences, it can be used to join with more BioBricks parts. Because of this property, the BioBricks assembly standard is said to be idempotent in nature. However, there will also be a "scar" sequence (either TACTAG or TACTAGAG) formed between the two fused BioBricks. This prevents BioBricks from being used to create fusion proteins, as the 6bp scar sequence codes for a tyrosine and a stop codon, causing translation to be terminated after the first domain is expressed, while the 8bp scar sequence causes a frameshift, preventing continuous readthrough of the codons. To offer alternative scar sequences that for example give a 6bp scar, or scar sequences that do not contain stop codons, other assembly standards such as the BB-2 Assembly, BglBricks Assembly, Silver Assembly and the Freiburg Assembly were designed.
While the easiest method to assemble BioBrick parts is described above, there also exist several other commonly used assembly methods that offer several advantages over the standard assembly. The 3 antibiotic (3A) assembly allows for the correct assembly to be selected via antibiotic selection, while the amplified insert assembly seeks to overcome the low transformation efficiency seen in 3A assembly.
The BioBrick assembly standard has also served as inspiration for using other types of endonucleases for DNA assembly. For example, both the iBrick standard and the HomeRun vector assembly standards employ homing endonucleases instead of type II restriction enzymes.
Type IIs restriction endonuclease assembly
Some assembly methods also make use of type IIs restriction endonucleases. These differ from other type II endonucleases as they cut several base pairs away from the recognition site. As a result, the overhang sequence can be modified to contain the desired sequence. This provides Type IIs assembly methods with two advantages – it enables "scar-less" assembly, and allows for one-pot, multi-part assembly. Assembly methods that use type IIs endonucleases include Golden Gate and its associated variants.
Golden Gate cloning

The Golden Gate assembly protocol was defined by Engler et al. 2008 to define a DNA assembly method that would give a final construct without a scar sequence, while also lacking the original restriction sites. This allows the protein to be expressed without containing unwanted protein sequences which could negatively affect protein folding or expression. By using the BsaI restriction enzyme that produces a 4 base pair overhang, up to 240 unique, non-palindromic sequences can be used for assembly.
Plasmid design and assembly
In Golden Gate cloning, each DNA fragment to be assembled is placed in a plasmid, flanked by inward facing BsaI restriction sites containing the programmed overhang sequences. For each DNA fragment, the 3' overhang sequence is complementary to the 5' overhang of the next downstream DNA fragment. For the first fragment, the 5' overhang is complementary to the 5' overhang of the destination plasmid, while the 3' overhang of the final fragment is complementary to the 3' overhang of the destination plasmid. Such a design allows for all DNA fragments to be assembled in a one-pot reaction (where all reactants are mixed together), with all fragments arranged in the correct sequence. Successfully assembled constructs are selected by detecting the loss of function of a screening cassette that was originally in the destination plasmid.
MoClo and Golden Braid
The original Golden Gate Assembly only allows for a single construct to be made in the destination vector . To enable this construct to be used in a subsequent reaction as an entry vector, the MoClo and Golden Braid standards were designed.
The MoClo standard involves defining multiple tiers of DNA assembly:
-
Tier 1: Tier 1 assembly is the standard Golden Gate assembly, and genes are assembled from their components parts (DNA parts coding for genetic elements like UTRs, promoters, ribosome binding sites or terminator sequences). Flanking the insertion site of the tier 1 destination vectors are a pair of inward cutting BpiI restriction sites. This allows these plasmids to be used as entry vectors for tier two destination vectors.The MoClo assembly standard allows for Golden Gate constructs to be further assembled in subsequent tiers. In the example here, four genes assembled via tier 1 Golden Gate assembly are assembled into a multi-gene construct in a tier 2 assembly.The Golden Braid assembly standard also builds on the first tier of Golden Gate assembly and assembles further tiers via a pairwise protocol. Four tier one destination vectors (assembled via Golden Gate assembly) are assembled into two tier 2 destination vectors, which are then used as tier 3 entry vectors for the tier 3 destination vector. Alternating restriction enzymes (BpiI for tier 2 and BsaI for tier 3) are used.The MoClo and Golden Braid assembly standards are derivatives of the original Golden Gate assembly standard.
- Tier 2: Tier 2 assembly involves further assembling the genes assembled in tier 1 assembly into multi-gene constructs. If there is a need for further, higher tier assembly, inward cutting BsaI restriction sites can be added to flank the insertion sites. These vectors can then be used as entry vectors for higher tier constructs.
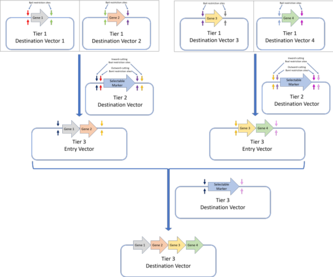
Each assembly tier alternates the use of BsaI and BpiI restriction sites to minimise the number of forbidden sites, and sequential assembly for each tier is achieved by following the Golden Gate plasmid design. Overall, the MoClo standard allows for the assembly of a construct that contains multiple transcription units, all assembled from different DNA parts, by a series of one-pot Golden Gate reactions. However, one drawback of the MoClo standard is that it requires the use of 'dummy parts' with no biological function, if the final construct requires less than four component parts. The Golden Braid standard on the other hand introduced a pairwise Golden Gate assembly standard.
The Golden Braid standard uses the same tiered assembly as MoClo, but each tier only involves the assembly of two DNA fragments, i.e. a pairwise approach. Hence in each tier, pairs of genes are cloned into a destination fragment in the desired sequence, and these are subsequently assembled two at a time in successive tiers. Like MoClo, the Golden Braid standard alternates the BsaI and BpiI restriction enzymes between each tier.
The development of the Golden Gate assembly methods and its variants has allowed researchers to design tool-kits to speed up the synthetic biology workflow. For example, EcoFlex was developed as a toolkit for E. Coli that uses the MoClo standard for its DNA parts, while a similar toolkit has also been developed for engineering the Chlamydomonas reinhardtii microalgae.
Site-specific recombination



Site-specific recombination makes use of phage integrases instead of restriction enzymes, eliminating the need for having restriction sites in the DNA fragments. Instead, integrases make use of unique attachment (att) sites, and catalyse DNA rearrangement between the target fragment and the destination vector. The Invitrogen Gateway cloning system was invented in the late 1990s and uses two proprietary enzyme mixtures, BP clonase and LR clonase. The BP clonase mix catalyses the recombination between attB and attP sites, generating hybrid attL and attR sites, while the LR clonase mix catalyse the recombination of attL and attR sites to give attB and attP sites. As each enzyme mix recognises only specific att sites, recombination is highly specific and the fragments can be assembled in the desired sequence.
Vector design and assembly
Because Gateway cloning is a proprietary technology, all Gateway reactions must be carried out with the Gateway kit that is provided by the manufacturer. The reaction can be summarised into two steps. The first step involves assembling the entry clones containing the DNA fragment of interest, while the second step involves inserting this fragment of interest into the destination clone.
- Entry clones must be made using the supplied "Donor" vectors containing a Gateway cassette flanked by attP sites. The Gateway cassette contains a bacterial suicide gene (e.g. ccdB) that will allow for survival and selection of successfully recombined entry clones. A pair of attB sites are added to flank the DNA fragment of interest, and this will allow recombination with the attP sites when the BP clonase mix is added. Entry clones are produced, and the fragment of interest is flanked by attL sites.
- The destination vector also comes with a Gateway cassette, but is instead flanked by a pair of attR sites. Mixing this destination plasmid with the entry clones and the LR clonase mix will allow for recombination to occur between the attR and attL sites. A destination clone is produced, with the fragment of interest successfully inserted. The lethal gene is inserted into the original vector, and bacteria transformed with this plasmid will die. The desired vector can thus be easily selected.
The earliest iterations of the Gateway cloning method only allowed for only one entry clone to be used for each destination clone produced. However, further research revealed that four more orthogonal att sequences could be generated, allowing for the assembly of up to four different DNA fragments, and this process is now known as the Multisite Gateway technology.
Besides Gateway cloning, non-commercial methods using other integrases have also been developed. For example, the Serine Integrase Recombinational Assembly (SIRA) method uses the ϕC31 integrase, while the Site-Specific Recombination-based Tandem Assembly (SSRTA) method uses the Streptomyces phage φBT1 integrase. Other methods, like the HomeRun Vector Assembly System (HVAS), build on the Gateway cloning system and further incorporate homing endonucleases to design a protocol that could potentially support the industrial synthesis of synthetic DNA constructs.

Long-overlap-based assembly
There have been a variety of long-overlap-based assembly methods developed in recent years. One of the most commonly used methods, the Gibson assembly method, was developed in 2009, and provides a one-pot DNA assembly method that does not require the use of restriction enzymes or integrases. Other similar overlap-based assembly methods include Circular Polymerase Extension Cloning (CPEC), Sequence and Ligase Independent Cloning (SLIC) and Seamless Ligation Cloning Extract (SLiCE). Despite the presence of many overlap assembly methods, the Gibson assembly method is still the most popular. Besides the methods listed above, other researchers have built on the concepts used in Gibson assembly and other assembly methods to develop new assembly strategies like the Modular Overlap-Directed Assembly with Linkers (MODAL) strategy, or the Biopart Assembly Standard for Idempotent Cloning (BASIC) method.
Gibson assembly
The Gibson assembly method is a relatively straightforward DNA assembly method, requiring only a few additional reagents: the 5' T5 exonuclease, Phusion DNA polymerase, and Taq DNA ligase. The DNA fragments to be assembled are synthesised to have overlapping 5' and 3' ends in the order that they are to be assembled in. These reagents are mixed together with the DNA fragments to be assembled at 50 °C and the following reactions occur:
- The T5 exonuclease chews back DNA from the 5' end of each fragment, exposing 3' overhangs on each DNA fragment.
- The complementary overhangs on adjacent DNA fragments anneal via complementary base pairing.
- The Phusion DNA polymerase fills in any gaps where the fragments anneal.
- Taq DNA ligase repairs the nicks on both DNA strands.
Because the T5 exonuclease is heat labile, it is inactivated at 50 °C after the initial chew back step. The product is thus stable, and the fragments assembled in the desired order. This one-pot protocol can assemble up to 5 different fragments accurately, while several commercial providers have kits to accurately assemble up to 15 different fragments in a two-step reaction. However, while the Gibson assembly protocol is fast and uses relatively few reagents, it requires bespoke DNA synthesis as each fragment has to be designed to contain overlapping sequences with the adjacent fragments and amplified via PCR. This reliance on PCR may also affect the fidelity of the reaction when long fragments, fragments with high GC content or repeat sequences are used.

MODAL
The MODAL strategy defines overlap sequences known as "linkers" to reduce the amount of customisation that needs to be done with each DNA fragment. The linkers were designed using the R2oDNA Designer software and the overlap regions were designed to be 45 bp long to be compatible with Gibson assembly and other overlap assembly methods. To attach these linkers to the parts to be assembled, PCR is carried using part-specific primers containing 15 bp prefix and suffix adaptor sequences. The linkers are then attached to the adaptor sequences via a second PCR reaction. To position the DNA fragments, the same linker will be attached to the suffix of the desired upstream fragment and the prefix of the desired downstream fragments. Once the linkers are attached, Gibson assembly, CPEC, or the other overlap assembly methods can all be used to assemble the DNA fragments in the desired order.
BASIC
The BASIC assembly strategy was developed in 2015 and sought to address the limitations of previous assembly techniques, incorporating six key concepts from them: standard reusable parts; single-tier format (all parts are in the same format and are assembled using the same process); idempotent cloning; parallel (multipart) DNA assembly; size independence; automatability.
DNA parts and linker design
The DNA parts are designed and cloned into storage plasmids, with the part flanked by an integrated prefix (iP) and an integrated suffix (iS) sequence. The iP and iS sequences contain inward facing BsaI restriction sites, which contain overhangs complementary to the BASIC linkers. Like in MODAL, the 7 standard linkers used in BASIC were designed with the R2oDNA Designer software, and screened to ensure that they do not contain sequences with homology to chassis genomes, and that they do not contain unwanted sequences like secondary structure sequences, restriction sites or ribosomal binding sites. Each linker sequence is split into two halves, each with a 4 bp overhang complementary to the BsaI restriction site, a 12 bp double stranded sequence and sharing a 21 bp overlap sequence with the other half. The half that is will bind to the upstream DNA part is known as the suffix linker part (e.g. L1S) and the half that binds to the downstream part is known as the prefix linker part (e.g. L1P). These linkers form the basis of assembling the DNA parts together.
Besides directing the order of assembly, the standard BASIC linkers can also be modified to carry out other functions. To allow for idempotent assembly, linkers were also designed with additional methylated iP and iS sequences inserted to protect them from being recognised by BsaI. This methylation is lost following transformation and in vivo plasmid replication, and the plasmids can be extracted, purified, and used for further reactions.
Because the linker sequence are relatively long (45bp for a standard linker), there is an opportunity to incorporate functional DNA sequences to reduce the number of DNA parts needed during assembly. The BASIC assembly standard provides several linkers embedded with RBS of different strengths. Similarly to facilitate the construction of fusion proteins containing multiple protein domains, several fusion linkers were also designed to allow for full read-through of the DNA construct. These fusion linkers code for a 15 amino acid glycine and serine polypeptide, which is an ideal linker peptide for fusion proteins with multiple domains.


Assembly
There are three main steps in the assembly of the final construct.
- First, the DNA parts are excised from the storage plasmid, giving a DNA fragment with BsaI overhangs on the 3' and 5' end.
- Next, each linker part is attached to its respective DNA part by incubating with T4 DNA ligase. Each DNA part will have a suffix and prefix linker part from two different linkers to direct the order of assembly. For example, the first part in the sequence will have L1P and L2S, while the second part will have L2P and L3S attached. The linker parts can be changed to change the sequence of assembly.
- Finally, the parts with the attached linkers are assembled into a plasmid by incubating at 50 °C. The 21 bp overhangs of the P and S linkers anneal and the final construct can be transformed into bacteria cells for cloning. The single stranded nicks are repaired in vivo following transformation, producing a stable final construct cloned into plasmids.
Applications
As DNA printing and DNA assembly methods have allowed commercial gene synthesis to become progressively and exponentially cheaper over the past years, artificial gene synthesis represents a powerful and flexible engineering tool for creating and designing new DNA sequences and protein functions. Besides synthetic biology, various research areas like those involving heterologous gene expression, vaccine development, gene therapy and molecular engineering, would benefit greatly from having fast and cheap methods to synthesise DNA to code for proteins and peptides. The methods used for DNA printing and assembly have even enabled the use of DNA as an information storage medium.
Synthesising bacterial genomes
Synthia and Mycoplasma laboratorium
On June 28, 2007, a team at the J. Craig Venter Institute published an article in Science Express, saying that they had successfully transplanted the natural DNA from a Mycoplasma mycoides bacterium into a Mycoplasma capricolum cell, creating a bacterium which behaved like a M. mycoides.
On Oct 6, 2007, Craig Venter announced in an interview with UK's The Guardian newspaper that the same team had synthesized a modified version of the single chromosome of Mycoplasma genitalium artificially. The chromosome was modified to eliminate all genes which tests in live bacteria had shown to be unnecessary. The next planned step in this minimal genome project is to transplant the synthesized minimal genome into a bacterial cell with its old DNA removed; the resulting bacterium will be called Mycoplasma laboratorium. The next day the Canadian bioethics group, ETC Group issued a statement through their representative, Pat Mooney, saying Venter's "creation" was "a chassis on which you could build almost anything". The synthesized genome had not yet been transplanted into a working cell.
On May 21, 2010, Science reported that the Venter group had successfully synthesized the genome of the bacterium Mycoplasma mycoides from a computer record, and transplanted the synthesized genome into the existing cell of a Mycoplasma capricolum bacterium that had its DNA removed. The "synthetic" bacterium was viable, i.e. capable of replicating billions of times. The team had originally planned to use the M. genitalium bacterium they had previously been working with, but switched to M. mycoides because the latter bacterium grows much faster, which translated into quicker experiments. Venter describes it as "the first species.... to have its parents be a computer". The transformed bacterium is dubbed "Synthia" by ETC. A Venter spokesperson has declined to confirm any breakthrough at the time of this writing.
Synthetic Yeast 2.0
As part of the Synthetic Yeast 2.0 project, various research groups around the world have participated in a project to synthesise synthetic yeast genomes, and through this process, optimise the genome of the model organism Saccharomyces cerevisiae. The Yeast 2.0 project applied various DNA assembly methods that have been discussed above, and in March 2014, Jef Boeke of the Langone Medical Centre at New York University, revealed that his team had synthesized chromosome III of S. cerevisiae. The procedure involved replacing the genes in the original chromosome with synthetic versions and the finished synthetic chromosome was then integrated into a yeast cell. It required designing and creating 273,871 base pairs of DNA – fewer than the 316,667 pairs in the original chromosome. In March 2017, the synthesis of 6 of the 16 chromosomes had been completed, with synthesis of the others still ongoing.