
Aziridines are organic compounds containing the aziridine functional group, a three-membered heterocycle with one amine (-NR-) and two methylene bridges (-CR
2-). The parent compound is aziridine (or ethylene imine), with molecular formula C
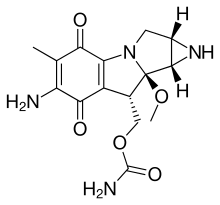
2H
4NH. Several drugs feature aziridine rings, including mitomycin C, porfiromycin, and azinomycin B (carzinophilin).
Structure
The bond angles in aziridine are approximately 60°, considerably less than the normal hydrocarbon bond angle of 109.5°, which results in angle strain as in the comparable cyclopropane and ethylene oxide molecules. A banana bond model explains bonding in such compounds. Aziridine is less basic than acyclic aliphatic amines, with a pKa of 7.9 for the conjugate acid, due to increased s character of the nitrogen free electron pair. Angle strain in aziridine also increases the barrier to nitrogen inversion. This barrier height permits the isolation of separate invertomers, for example the cis and trans invertomers of N-chloro-2-methylaziridine.
Synthesis
Several routes have been developed for the syntheses of aziridines (aziridination).
Cyclization of haloamines and amino alcohols
An amine functional group displaces the adjacent halide in an intramolecular nucleophilic substitution reaction to generate an aziridine. The parent aziridine is produced industrially from aminoethanol via two related routes. The Nippon Shokubai process requires an oxide catalyst and high temperatures to effect the dehydration. In the Wenker synthesis, the aminoethanol is converted to the sulfate ester, which undergoes base-induced sulfate elimination.
Nitrene addition
Nitrene addition to alkenes is a well-established method for the synthesis of aziridines. Photolysis or thermolysis of organic azides are good ways to generate nitrenes. Nitrenes can also be prepared in situ from iodosobenzene diacetate and sulfonamides, or the ethoxycarbonylnitrene from the N-sulfonyloxy precursor.

From triazolines, epoxides, and oximes
Thermolysis or photolysis of triazolines expels nitrogen, producing an aziridine. One method involves the ring-opening reaction of an epoxide with sodium azide, followed by reduction of the azide with triphenylphosphine accompanied by expulsion of nitrogen gas:

Another method involves the ring-opening reaction of an epoxide with amines, followed by ring closing with the Mitsunobu reaction.
The Hoch-Campbell ethylenimine synthesis involves the reaction of certain oximes with Grignard reagents, which affords aziridines.

From alkenes using DPH
Aziridines are obtained by treating a mono-, di-, tri- or tetra- substituted alkene (olefin) with O-(2,4-dinitrophenyl)hydroxylamine (DPH) in the presence of rhodium catalysis.
Alkene + DPH Aziridine

For instance, Ph-Aziridine-Me can be synthesized by this method and then converted by ring opening reaction to (D)-amphetamine and (L)-amphetamine (the two active ingredients in Adderall).
From α-chloroimines
The De Kimpe aziridine synthesis allows for the generation of aziridines by reacting an α-chloroimine with a nucleophile, such as hydride, cyanide, or a Grignard reagent.
From 2-azido alcohols
2-azido alcohols can be converted into aziridines with the use of trialkyl phosphines such as trimethylphosphine or tributylphosphine.
Reactions
Nucleophilic ring opening
Aziridines are reactive substrates in ring-opening reactions with many nucleophiles due to their ring strain. Alcoholysis and aminolysis are basically the reverse reactions of the cyclizations. Carbon nucleophiles such as organolithium reagents and organocuprates are also effective.
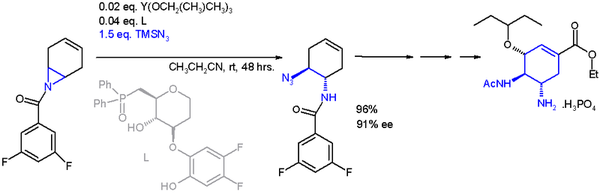
One application of a ring-opening reaction in asymmetric synthesis is that of trimethylsilylazide TMSN
3 with an asymmetric ligand in scheme 2 in an organic synthesis of oseltamivir:
1,3-dipole formation
Certain N-substituted azirines with electron withdrawing groups on both carbons form azomethine ylides in an electrocyclic thermal or photochemical ring-opening reaction. These ylides can be trapped with a suitable dipolarophile in a 1,3-dipolar cycloaddition.

When the N-substituent is an electron-withdrawing group such as a tosyl group, the carbon-nitrogen bond breaks, forming another zwitterion TsN−
–CH
2–CH+
2–R

This reaction type requires a Lewis acid catalyst such as boron trifluoride. In this way 2-phenyl-N-tosylaziridine reacts with alkynes, nitriles, ketones and alkenes. Certain 1,4-dipoles form from azetidines.
Other
N-unsubstituted aziridines can be opened with olefins in the presence of strong Lewis acid B(C
6F
5)
3.
Safety
As electrophiles, aziridines are subject to attack and ring-opening by endogenous nucleophiles such as nitrogenous bases in DNA base pairs, resulting in potential mutagenicity.
The International Agency for Research on Cancer (IARC) classifies aziridine compounds as possibly carcinogenic to humans (IARC Group 2B). In making the overall evaluation, the IARC Working Group took into consideration that aziridine is a direct-acting alkylating agent, which is mutagenic in a wide range of test systems and forms DNA adducts that are promutagenic. The features that are responsible for their mutagenicity are relevant to their beneficial medicinal properties.
See also
- Binary ethylenimine, a dimeric form of aziridine