
Bcr-Abl tyrosine-kinase inhibitors (TKI) are the first-line therapy for most patients with chronic myelogenous leukemia (CML). More than 90% of CML cases are caused by a chromosomal abnormality that results in the formation of a so-called Philadelphia chromosome. This abnormality was discovered by Peter Nowell in 1960 and is a consequence of fusion between the Abelson (Abl) tyrosine kinase gene at chromosome 9 and the break point cluster (Bcr) gene at chromosome 22, resulting in a chimeric oncogene (Bcr-Abl) and a constitutively active Bcr-Abl tyrosine kinase that has been implicated in the pathogenesis of CML. Compounds have been developed to selectively inhibit the tyrosine kinase.
Before the 2001 U.S. Food and Drug Administration (FDA) approval of imatinib, no drugs were available to alter the natural progression of CML. Only cytotoxic drugs such as busulfan, hydroxyurea or interferon-alpha (rIFN-α) were utilized. Even though the first Bcr-Abl TK inhibitor was named "the magic bullet" to cure cancer by Time magazine, a second generation of Bcr-Abl TKI was subsequently developed to combat the initial resistance that emerged.
New forms of resistance can arise as: missense mutations within the Abl kinase domain, over-expression of Bcr-Abl, increased production of transmembrane plasma proteins, or the constitutive activation of downstream signaling molecules such as Src-family kinases.
History
CML has a well defined molecular target and relatively selective therapies aimed at that target, which is not the case for the majority of cancers and chemotherapies today. Bcr-Abl was regarded as highly attractive target for drug intervention since the Bcr-Abl fusion gene encodes a constitutively activated kinase. Drug discovery that specifically targeted the ATP binding site of a single kinase was regarded as quite a challenging task since hundreds of protein kinases were known in the human genome. In the presence of TKI the binding of ATP is blocked, phosphorylation is prevented and Bcr-Abl expressing cells either have a selective growth disadvantage or undergo apoptotic cell death.
Due to increasing resistance and intolerance to imatinib efforts were made to develop new drugs that could inhibit the Bcr-Abl tyrosine kinase. This led to the discovery of second generation drugs. While drug screening was used to develop imatinib, second generation TKI's were developed with rational drug design approach due to increased knowledge in structural biology of the Bcr-Abl tyrosine kinase.
First generation
Imatinib (STI571)
Imatinib (Gleevec) was discovered in 1992 and is regarded as first generation drug since it is the first Bcr-Abl tyrosine kinase inhibitor to be used in the treatment of CML.
Development

In the development of imatinib, the structure of Bcr-Abl tyrosine kinase played a limited role because it was unknown. A high-throughput screening of chemical libraries at Novartis was performed to identify a starting molecule, which was called "Pyrimidine A". This compound served as a lead compound and was then tested and modified to develop imatinib. With a replacement of the imidazole group with a benzamido group, the compound's specificity increased while its activity as a kinase inhibitor remained the same. Subsequently, introducing a methyl substituent ortho to the pyrimidinyl-amino group enhanced the potency.
Binding

Since then crystallographic studies have revealed that imatinib binds to the kinase domain of Abl only when the domain adopts the inactive or "closed" conformation. This is where the glycine-rich, P-binding phosphate loop (P-loop) folds over the ATP binding site and the activation-loop adopts a conformation in which it occludes the substrate binding site and disrupts the ATP phosphate binding site to block the catalytic activity of the enzyme. The shift of the AspPheGly (DFG) triad at the N-terminal end of the activation loop results in the exposure of a binding pocket which can be utilized by inhibitors. This conformation is referred to as DFGout.
Imatinib binds to Abl domain via six hydrogen bond interactions. This stabilizes the imatinib Bcr-Abl complex and prevents ATP from reaching its binding site. The hydrogen bonds involve the pyridine-N and backbone-NH of Met-318, the aminopyrimidine and side chain hydroxyl of Thr-315, the amide-NH and side chain carboxylate of Glu-286, the carbonyl and backbone-NH of Asp-381, the protonated methylpiperazine with the backbone-carbonyl atoms of Ile-360 and His-361. Additionally, a number of van der Waals interactions contribute to binding. A hydrophobic pocket is formed by amino acid residues Ile-293, Leu-298, Leu-354 and Val-379 around the phenyl ring adjacent to the piperazinyl-methyl group of imatinib. At the time of its discovery, in the absence of structural information, no clear explanation for the impressive selectivity of imatinib could be found.
Although first-generation treatment achieved an extremely high response rate and a low relapse rate in CML patients, some patients do experience resistance or intolerance to imatinib.
Drug resistance
Drug resistance is the main drive in continuing research and development of Bcr-Abl TKI. Shortly after the introduction of imatinib, investigators began to describe a number of in vitro derived cell lines with resistance to the drug. This was rapidly followed by the clinical description of imatinib resistant cells in patients, which has resulted in efforts to better understand the biology behind these observations. Assessments of therapeutic response of imatinib in patients with CML are based upon meeting hematologic, cytogenic and molecular milestones. Patients that fail to achieve defined responses at predefined time points are described as primarily resistant to therapy, and those losing previously obtained milestones in disease regression are termed secondarily resistant. Before a conclusion is drawn, it is important to consider that retrospective data has showed a high incidence of imatinib non-compliance in CML patients and this could lead to undesired clinical outcomes.

In general, imatinib resistance can be subdivided into Bcr-Abl dependent and independent mechanisms. Bcr-Abl dependent mechanisms include over expression or amplification of the Bcr-Abl gene and point mutations within the Bcr-Abl kinase domain that interfere with imatinib binding. Bcr-Abl independent mechanisms include factors influencing the concentration of imatinib within the cell, for example by alterations in drug influx and efflux and activation of Bcr-Abl independent pathways, such as members of the Src kinase family. Imatinib resistance can also be produced by other mechanisms that will not be mentioned here as the importance of those mechanisms still remain a question due to lack of clinical data.
Bcr-Abl dependent mechanisms of resistance
Bcr-Abl duplication
The first reports of resistance to imatinib described a development of oncogene amplification. That is, the gene that encodes for the pathogenic Bcr-Abl tyrosine kinase is duplicated in the DNA sequence, leading to higher expression of the pathogen. Increasing the imatinib dose could surmount this kind of resistance, provided that severe or intolerable adverse effects are not produced.
Bcr-Abl mutation
Point mutations can cause amino acid substitutions inside the kinase domain of the Bcr-Abl protein and disrupt the binding site of imatinib on the tyrosine kinase, resulting in a loss of sensitivity to the drug. These mutations normally affect the structure of the Bcr-Abl protein, leading either to interruption of critical contact points between the drug and the Bcr-Abl protein or induction of a conformational change, resulting in a protein that imatinib is unable to bind to.
Mutational frequencies appear to increase as the disease, CML, progresses from chronic phase to the blast phase. The most important mutations are the P-loop mutations and the T315I mutation. Mutations on other sites of the kinase have also been reported, for example on the C-helix, SH2 domain, substrate binding site, activation loop and C-terminal lobe. Some of these mutations have clinical significance, but none as much as P-loop and T315I mutations.
T315I mutation
The T315I is a unique mutation because of its resistance to all approved Bcr-Abl inhibitors, prior to ponatinib. It is caused by a single cytosine to thymine (C -> T) base pair substitution at position 944 of the Abl gene (codon '315' of the Abl protein) sequence resulting in amino acid Threonine being substituted by Isoleucine at that position - thus 'T315I'. This substitution eliminates a critical oxygen molecule needed for hydrogen bonding between imatinib and the Abl kinase, and also creates steric hindrance to the binding of most TKIs. When discovered, it was estimated that every 6 out of 9 cases of advanced stage CML with imatinib resistance carried this mutation. T315I produces the highest magnitude of resistance of any mutation both to imatinib and second generations TKIs.Ponatinib (Iclusig) by Ariad was approved in 2013 for use as second-line CML treatment, and is the only licensed TKI which binds to the T315I mutated kinase successfully.
P-loop mutations
The structure of Bcr-Abl contains two flexible loops, the ATP-binding P-loop and the activation loop. These loops have specific arrangements in the inactive conformation of Bcr-Abl that stabilize the basal conformation. Mutations in these loops destabilize arrangement of the loops such that the kinase domain cannot assume the inactive conformation required for imatinib binding. Mutations in the P-loop region are the most common, accounting for 36-48% of all mutations. There are clinical data indicating that Bcr-Abl mutations in the P-loop is 70-100 fold less sensitive to imatinib compared with native Bcr-Abl.
Bcr-Abl Independent mechanisms of resistance
Additional mechanisms have been postulated to describe resistance seen in various model systems although none have been clearly identified as a sole source of clinical resistance.
Drug efflux caused by P-glycoproteins
Some investigations in cell lines have shown that imatinib resistance may be partly due to an increase in the expression of the P-glycoprotein efflux pump. By utilizing agents that inhibit P-glycoprotein activity imatinib susceptibility has been restored in some cases.
Drug import by organic cation transporter 1
The entry of imatinib into cells is dependent on an organic cation transporter (OCT1). OCT1 plays a significant role in imatinib resistance by inhibiting its influx and thus decreasing the intracellular bioavailability of imatinib. Patients with low expression, activity or polymorphisms of OCT1 had significantly lower intracellular levels of imatinib. The response of patients with low OCT1 activity was significantly dose-dependent. This data indicates that OCT1 activity is an important determinant in the molecular response to imatinib.
Alternative signaling pathway activation
In a few patient groups, resistance may be caused by the activation of other signaling pathways, particularly the Src family kinases. The Src family kinases have been implicated in Bcr-Abl signaling and mediate imatinib resistance by stabilizing the active conformation of Bcr-Abl, a conformation that does not bind imatinib. Furthermore, increasing evidence suggests that Src family kinases are also involved in Bcr-Abl-independent forms of imatinib resistance.
Solutions
The treatment options for imatinib resistant or intolerant CML patients may include strategies such as increasing the dose of imatinib or the use of second-generation drugs. Escalation of imatinib-doses has shown to overcome some cases of primary resistance to imatinib, such as Bcr-Abl duplication, but the response is usually short acting. In the case of resistance or intolerance, it could be helpful to test for Bcr-Abl mutations to direct the choice of second line treatment as the variable options have different function profile against the different mechanisms of resistance. Second-generation drugs offer improved potency and a greater likelihood of success in resistant patients. There is also a growing interest in testing the hypothesis that administration of multiple Abl kinase inhibitors in early phase patients could be used to delay or prevent the emergence of drug resistant clones. The combination of two agents targeting different pathways involved in CML may significantly improve response rates and potentially increase survival.
Second generation drugs
Second generation drugs are intended to have decreased resistance and intolerance than imatinib. Second generation drugs that are currently marketed are nilotinib, dasatinib, bosutinib and ponatinib.
Nilotinib (AMN107)

Development
Nilotinib is a phenylamino-pyrimidine derivative that is structurally related to imatinib. It was developed based on the structure of the Abl-imatinib complex to address the need associated with imatinib intolerance and resistance. Small changes were made on the imatinib molecule to make it more potent and selective as a Bcr-Abl inhibitor and these changes resulted in the discovery of nilotinib. Nilotinib is a selective Bcr-Abl kinase inhibitor.
Nilotinib is 10-30 fold more potent than imatinib in inhibiting activity of the Bcr-Abl tyrosine kinase and proliferation of Bcr-Abl expressing cells. The drug effectively inhibits the auto phosphorylation of Bcr-Abl on Tyr-177 that is involved in CML pathogenesis.Synergistic activity of imatinib and nilotinib has been reported following coadministration. This might be a result of the fact that the drugs are taken up in cells by different mechanisms: imatinib influx is dependent on OCT1 but nilotinib is not. Nilotinib is also not a substrate for the efflux transporter P-glycoprotein pump, unlike imatinib. Although the two dimensional molecular structures of these two drugs might look similar, they are dissimilar in terms of spatial structure and molecular properties.

Binding
Nilotinib binds to the inactive conformation of the Abl kinase domain, largely through lipophilic interactions and thus blocks its catalytic activity. Nilotinib binds to the kinase domain by making four hydrogen bond interactions involving the pyridyl-N and the backbone NH of Met-318, the anilino-NH and the side chain OH of Thr-315, the amido-NH and side chain carboxylate of Glu-286 and the amido carbonyl with the backbone NH of the Asp-381. The [4-(3-pyridinyl)-2-pyrimidinyl] anilino segment of nilotinib has close binding interactions with Met-318, Phe-317 and Thr-315 residues of a region within the ATP binding site. The remaining half of the compound extends beyond the Thr-315 gatekeeper residue to bind within an additional pocket. The 3-methylimidazole and trifluoro-methyl groups of nilotinib make important interactions with the Abl kinase domain. These groups also make the shape of nilotinib very different from that of imatinib. Nilotinib also binds to the kinase through a large number of weak van der Waals interactions.
Resistance
Nilotinib has shown effect against most mutations (32/33) that are associated with imatinib resistance but the T315I mutant remains resistant to nilotinib. Its ineffectiveness against the T315I mutant seems to be a consequence of the loss of an H-bond interaction between threonine-O and aniline-NH on nilotinib and a steric clash between the isoleucine-methyl group and 2-methylphenyl phenyl group of nilotinib. On the other hand, resistance to nilotinib is associated with a limited spectrum of Bcr-Abl kinase mutations that mostly affect the P-loop and T315I. However all mutations except T315I were effectively suppressed by increasing nilotinib concentration. Although nilotinib is more potent than imatinib it is possible that its specific mode of binding to Abl may make other sites vulnerable to new kinds of drug resistance.
Dasatinib (BMS-354825)

Development
Dasatinib is a thiazolylaminopyrimidine developed as the hydrochloride salt. It was discovered with a program directed towards immunosuppressive drugs and is 325-fold more potent against cells expressing wild type Bcr-Abl than imatinib. Dasatinib is a multi targeted inhibitor of Bcr-Abl and Src family kinases. It also has inhibitory activity against additional downstream kinases.

Binding
Dasatinib binds to Abl with less stringent conformational requirements than imatinib so it exhibits increased potency but reduced selectivity compared to imatinib. Dasatinib binds both the active and inactive conformation of Abl kinase, contrary to the binding of most other TKIs to the active form only. Compounds that target the active conformation have been identified but the binding site in all the hundreds of human protein kinases is very similar. Therefore, there is a considerably greater scope for dissimilarities between the inactive conformations so the efforts to discover highly selective kinase inhibitors are being directed towards molecules that bind to the inactive conformation.
Dasatinib has some structural elements in common with nilotinib, in particular the juxtaposition of the aminopyrimidine and the carboxamide groups. The aminothiazole segment of dasatinib makes a bi-dentate H-bonding interaction with the backbone CO and NH of Met-318 and the amide-NH makes an H-bond with the side chain oxygen of Thr-315.
Resistance
Since dasatinib is an inhibitor of Src family kinases, it can overcome resistance due to Src family kinase activation. Because it does not bind to Bcr-Abl with the same stringent conformational requirements as imatinib, it can inhibit all Bcr-Abl kinase domain mutants except for T315I. Dasatinib is also not a substrate of multidrug P-glycoprotein efflux pumps like imatinib. Because of this dasatinib may be active in some patients after failure with both imatinib and nilotinib. Although dasatinib is much more potent than imatinib it is possible, like with nilotinib, that its specific mode of binding to Abl may lead to new vulnerable sites that could confer new kinds of drug resistance. Mutations have been found on Phe317 so that is a potential vulnerable site for this drug.
Bosutinib (SKI-606)
Development

Bosutinib's structure is based on a quinoline scaffold and is structurally related to the AstraZeneca quinazoline template. Src kinase dependent yeast screening led to characterization of a 4-anilino-3-quinolinecarbonitrile as an Src inhibitor. Combination of the features of this hit and a related compound, and attachment of solubilizing groups, led to the discovery of bosutinib. It was suggested to be an Abl kinase inhibitor and when tested as such it turned out to be slightly more potent against Abl than Src (IC50 1,4 nM vs. 3,5 nM). Bosutinib's activity was first described in 2001 and it was disclosed as an Abl kinase inhibitor in 2003. At first it was believed that bosutinib was a selective Src kinase inhibitor but now it is known that its kinase inhibition profile is far less restricted than originally thought. Bosutinib inhibits Src, Abl and a wide range of both tyrosine and serine-threonine kinases.
Resistance
Bosutinib inhibited cells expressing a variety of mutations, some of which led to imatinib resistance, but the T315 mutation was completely resistant to bosutinib. In contrast to imatinib, nilotinib and dasatinib, bosutinib is not an efficient substrate for multidrug resistance (MDR) transporters that promotes efflux of foreign molecules from cells. Bosutinib even inhibits these transporter proteins in higher concentrations.
Ponatinib (AP24534)
ARIAD Pharmaceuticals, Inc. announced on September 10, 2010 that ponatinib, an orally active Bcr-Abl TKI effective against the T315I mutation had been approved for a phase II clinical trial.
The road to discovery can be linked to AP23464, one of the first of Ariad's ATP competitive dual Src/Abl inhibitors. AP23464 was identified using structure base drug design and focused synthetic libraries of trisubstituted purine analogs. The substance potently inhibits, on nanomolar scale, Src and Bcr-Abl kinases including many common imatinib resistant Bcr-Abl mutations. AP23464 does not inhibit the T315I mutation, however, whereas AP24534 (ponatinib) does.
Development

Ariad used the highly potent drug lead, AP23464 to further investigate inhibitory possibilities of purine cored templates for dual Src/Abl inhibitors. First, searching for substances effective on the inactive conformation of Abl, the side chain bound to the nitrogen on the purine core was replaced with a diarylamide structure, that was known to have a high affinity to the inactive conformation by forming crucial hydrogen bonds and filling hydrophobic pockets on the kinase. Furthermore, it was determined that the cyclopentyl group on the purine core clashed with a glycine rich P-loop in that confirmation and was thus removed from the molecule. Then with in-vitro testing on inhibitory activity and in-vivo oral absorption assays a more lipophilic, amide bound, cyclopropyl group on C6 on the purine core was found to display both satisfactory pharmacokinetics and efficacy. Finally modifications on the diarylamide side chain by adding imidazole appendages were inspired by then newly released nilotinib structure. Those modifications resulted in what was called AP24163. During this development cycle, Ariad tested several substances against cells transfected with T315I mutated Bcr-Abl kinase and, surprisingly, found AP24163 demonstrated reasonable inhibitory action on top of potent inhibition of native Bcr-Abl.
Following up on that breakthrough Ariad began further research to increase the efficacy of compound AP24163 against the T315I mutation. Docking of the molecule into the ATP binding site of T315I mutated Bcr-Abl kinase revealed that the expected steric clash with isoleucine was not present due to a lesser sterically demanding vinyl linkage between the purine core and the diarylamide side chain compared to other TKIs. The first step was to try to find an even less sterically demanding structure. First an acetylene linkage was tested, that resulted in higher potency but unfavorable pharmacokinetics. Later, a more stable 2-butyne linkage was selected. To achieve this linkage an imidazol[1,2-a]pyridine core was used as a starting material for a Sonogashira reaction; but the pharmacokinetics were still poor. When developing AP24163, adding a cyclopropane side chain on C8 in the purine core resulted in favorable pharmacokinetics. Several different side chains were then tested, but the best results were obtained with no side chain at all, resulting a substance with satisfactory pharmacokinetics, but now with reduced potency against T315I also. The first step in increasing the potency again was to look at other TKI's. Imatinib has a terminal methyl piperazine group which has been shown to form a hydrogen bond with the carbonyl oxygen atom of residue Ile-360 in the activation loop of the Abl kinase. The piperazine ring is also a common solubilizing group that could further improve the pharmacokinetic properties of the molecule. Those speculations were confirmed with a two-fold increase in inhibitory action against Bcr-Abl T315I mutated kinase and the silver lining was the plasma protein binding of the substance (named '19a') appeared to have decreased, allowing for smaller doses with the same potency. Whilst '19a' exhibited good oral pharmacokinetics in both mice and rats, it also retained high partition coefficient at 6.69. So, in attempts to reduce the molecule's lipophilicity further, substitution of a single carbon atom on the imidazo[1,2-a]pyridine core was made; which resulted in what is now known as the compound ponatinib.
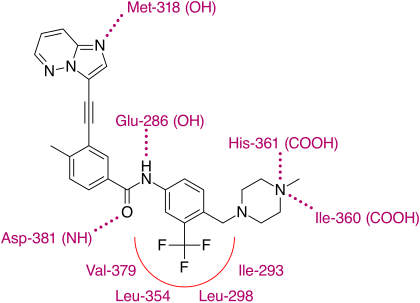
Binding
X-ray crystallographic analysis of ponatinib and T315I Bcr-Abl mutated kinase display that the imidazo[1,2b]pyridazine core rests in the adenine pocket of the enzyme. The methylphenyl group occupies a hydrophobic pocket behind I315, the ethynyl linkage forms favorable van der Waals interactions with the amino acid and the trifluoromethyl group binds to a pocket induced by the inactive conformation kinase. Also in the conformation of the kinase that ponatinib rests in, additional favorable van der Waals interactions between the drug and Tyr-253 and Phe-382. Five hydrogen bonds are generated, with the backbone of Met-318 in the hinge region, with the backbone of Asp-381, with the side chain of Glu-286 and the protonated methylpiperazine with the backbone-carbonyl atoms of Ile-360 and His-361.
With this structure ponatinib has been shown to have a relatively broad kinase specificity profile which can probably be linked to the linearity of the linkage section of the molecule. With this linear structure the drug appears to avoid steric clashes with hydrophobic TK gatekeeper residues. Despite, or even because of this, ponatinib is a potent drug and targets not just most of the known mutations on the Bcr-Abl TK but, most importantly of all, T315I. This mutation is emerging as a common pathway to failure of both first and second line treatments. Unlike other T315I targeting inhibitors in development, ponatinib does not target Aurora kinases, which clearly distinguishes it from them and emphasizes the significance of its discovery.
Bafetinib (INNO-406)
With the emerging resistance to imatinib treatment after its launch alternative treatment was highly sought after. Bafetinib was the offspring of an attempt to create a more potent drug than imatinib, with efficacy against various point mutations in the Bcr-Abl kinase, with fewer adverse effects and with narrower kinase spectra, namely just Lyn and Bcr-Abl.
Development
In the search for a substance that fit the criteria mentioned, the crystal structure of imatinib bound to Abl was examined. This revealed a hydrophobic pocket around the phenyl ring adjacent to the piperazinylmethyl group of imatinib. Attempts to utilize this pocket to increase efficacy led to the addition of various hydrophobic groups including single fluoro, bromo and chloro substituents. Finally a trifluoromethyl group at position 3 was found to give the best results, with approximately 36-fold improvement over imatinib. The addition of a hydrophobic group now needed to be countered to sustain the solubility of the substance. Closer examination of the crystal structure of imatinib-kinase complex revealed Tyr-236 was in close proximity to the pyridine ring of imatinib, suggesting there was little or no room for a larger group there. With that in mind a more hydrophilic pyrimidine ring was substituted for the pyridine, which was found to increase solubility while leaving efficacy the same or even slightly greater. Finally to improve the hydrogen bonding of the piperazine ring of imatinib with Ile-360 and His-361, pyrrolidine and azetidine derivatives were introduced. The most promising substance from these final modifications was labeled NS-187.
Binding

Due to the structural similarities of imatinib and bafetinib, their binding to Bcr-Abl is also quite similar. The only notable difference comes from the hydrophobic interaction between the trifluoromethyl group and the hydrophobic pocket created by Ile-293, Leu-298, Leu-354, and Val-379. This group can also be linked to bafetinib's specificity for Lyn, as the binding site there is almost identical to that on Bcr-Abl.
Bafetinib has its place in TKI therapy as it is effective both against most imatinib resistant mutations (not including T315I) and some dasatinib resistant mutations. Bafetinib also has more affinity for Bcr-Abl than nilotinib (but less than dasatinib) but only targets Bcr-Abl and Src family kinases Lck and Lyn; with unrivalled specificity which suggests the probability of fewer adverse effects.
CytRx has bafetinib in phase II clinical trial as a treatment for leukemia as of May 2010.
1,3,4 thiadiazole derivatives - Substance 14
Some interest has been with thiazol and thiadiazole derivatives and their ability to inhibit Bcr-Abl TKs.
Development

One Italian research group discovered through digital screening that commercially available thiadiazole derivatives displayed moderate inhibitory action on both Abl and Src kinases. Using a 1,3,4 thiadiazol core and trying different groups or molecules on the benzene rings, several different substances with inhibitory properties were produced. The flexibility of the core allowed a number of conformations of the substances to bind to the ATP site of the Abl kinase, though all of them bound to the kinase's active form. Further study of the binding showed that the position of the sulfur that binds to the toluene structure played an important role in regard to Abl binding and also that only one of the nitrogen's one thiadiazole formed a hydrogen bond. Furthermore, computer analysis of the structure showed the amide connected benzene-ketone could be substituted for a more favorable thiophene ring. Though it has to be noted this analysis was done with comparing the crystal structure of Abl and dasatinib, which is the inactive conformation of Abl, the knowledge gathered from the docking and structure analysis led to identification of a compound, referred to as substance 14, with a high affinity to Abl.
Binding
The binding of substance 14 is partly similar to dasatinib, the aminothiazole segment of substance 14 makes a bi-dentate H-bonding interaction with the backbone CO and NH of Met-318 while the methoxy-benzene falls nicely into a hydrophobic pocket created by Val 256, Ala 253, Lys 271 and Ala 380. Whilst the similar binding properties to those of dasatinib, suggests the possibility of producing Bcr-Abl TKI's from thiazole cores is real, the question remains open whether this research will just lead to a dasatinib analog or a novel way to inhibit TKs.
Others
Rebastinib (DCC-2036) Also an inhibitor of TIE-2 and VEGFR-2. It has had a phase 1 clinical trial for Leukemias (Ph+ CML With T315I Mutation). It is in a phase 1 clinical trial of combination therapy for metastatic breast cancer.
Asciminib (ABL001) is an inhibitor of the Abelson kinase targeting the myristoyl pocket to allosterically inhibit the enzyme. As of August 2020, it had completed a phase III study in CML (ASCEMBL) showing superior efficacy to bosutinib.
Summary
| Drug | Structure | H-bonds | H-bonding amino acids | Binding confirmation | Discovery | Status as of 2017 |
|---|---|---|---|---|---|---|
| Imatinib (STI571) |
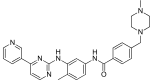
|
6 | Met-318, Thr-315, Glu-286, Asp-381, Ile-380, His-361 | Inactive | Drug screening | Marketed as first line therapy |
| Nilotinib (AMN107) |

|
4 | Met-318, Thr-315, Glu-286, Asp-381 | Inactive | Rational drug design | Marketed as second line therapy |
| Dasatinib (BMS-345825) |

|
3 | Met-318, Thr-315 | Active | Rational drug design | Marketed as second line therapy |
| Bosutinib (SKI-606) |

|
- | - | Inactive | Rational drug design | Marketed as second line therapy |
| Ponatinib (AP-24534) |

|
5 | Met-318, Asp-381, Glu-286, His-381, Ile-380 | Inactive | Rational drug design | Marketed as second line therapy |
| Bafetinib (INNO-406) |
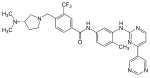
|
6 | Met-318, Thr-315, Glu-286, Asp-381, His-361, Ile-360 | Inactive | Rational drug design | Marketed as second line therapy |
Current status - re Ph+ CML
Imatinib remains a standard frontline TKI. Nilotinib and dasatinib are also approved by the FDA as frontline drugs, in June and October 2010, respectively. Four of these drugs, nilotinib, dasatinib, bosutinib and ponatinib are approved for the treatment of imatinib-resistant or intolerant CML. The first-line data for these compounds are encouraging and suggest that some or all of them may replace imatinib as a frontline standard TKI in the future.