
A biosimilar (also known as follow-on biologic or subsequent entry biologic) is a biologic medical product that is almost an identical copy of an original product that is manufactured by a different company. Biosimilars are officially approved versions of original "innovator" products and can be manufactured when the original product's patent expires. Reference to the innovator product is an integral component of the approval.
Unlike with generic drugs of the more common small-molecule type, biologics generally exhibit high molecular complexity and may be quite sensitive to changes in manufacturing processes. Despite that heterogeneity, all biopharmaceuticals, including biosimilars, must maintain consistent quality and clinical performance throughout their lifecycle.
Drug-related authorities such as the European Medicines Agency (EMA) of the European Union, the United States Food and Drug Administration (FDA), and the Health Products and Food Branch of Health Canada hold their own guidance on requirements for demonstration of the similar nature of two biological products in terms of safety and efficacy. According to them, analytical studies demonstrate that the biological product is highly similar to the reference product, despite minor differences in clinically inactive components, animal studies (including the assessment of toxicity), and a clinical study or studies (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics). They are sufficient to demonstrate safety, purity, and potency in one or more appropriate conditions of use for which the reference product is licensed and is intended to be used and for which licensure is sought for the biological product.
The World Health Organization (WHO) published its "Guidelines for the evaluation of similar biotherapeutic products (SBPs)" in 2009. The purpose of this guideline is to provide an international norm for evaluating biosimilars.
The EMA has granted marketing authorizations for more than 50 biosimilars since 2006, (first approved biosimilar Somatropin (Growth hormone)). The first biosimilar of a monoclonal antibody to be approved worldwide was a biosimilar of infliximab in the EU in 2013. On March 6, 2015, the FDA approved the United States' first biosimilar product, the biosimilar of filgrastim called filgrastim-sndz (trade name Zarxio) by Sandoz.
Approval processes
United States
In the United States, the Food and Drug Administration (FDA) held that new legislation was required to enable them to approve biosimilars to those biologics originally approved through the PHS Act pathway. Additional Congressional hearings have been held. On March 17, 2009, the Pathway for Biosimilars Act was introduced in the House. See the Library of Congress website and search H.R. 1548 in 111th Congress Session. Since 2004 the FDA has held a series of public meetings on biosimilars.
The FDA gained the authority to approve biosimilars (including interchangeables that are substitutable with their reference product) as part of the Patient Protection and Affordable Care Act signed into law by President Obama on March 23, 2010.
The FDA has previously approved biologic products using comparability, for example, Omnitrope in May 2006, but this like Enoxaparin was also to a reference product, Genotropin, originally approved as a biologic drug under the FD&C Act.
On March 6, 2015, Zarxio obtained the first approval of FDA. Sandoz's Zarxio is biosimilar to Amgen's Neupogen (filgrastim), which was originally licensed in 1991. This is the first product to be passed under the Biologics Price Competition and Innovation Act of 2009 (BPCI Act), which was passed as part of the Affordable Healthcare Act. But Zarxio was approved as a biosimilar, not as an interchangeable product, the FDA notes. And under the BPCI Act, only a biologic that has been approved as an "interchangeable" may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product. The FDA said its approval of Zarxio is based on review of evidence that included structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data and other clinical safety and effectiveness data that demonstrates Zarxio is biosimilar to Neupogen.
In March 2020, most protein products that were approved as drug products (including every insulin currently on the market as of December 2019) are scheduled to open up to biosimilar and interchangeable competition in the United States. However, "chemically synthesized polypeptides" are excluded from this transition, which means that a product that falls within this category won't be able to come to market as a biosimilar or interchangeable product, but will have to come to the market under a different pathway.
Background
Cloning of human genetic material and development of in vitro biological production systems has allowed the production of virtually any recombinant DNA based biological substance for eventual development of a drug. Monoclonal antibody technology combined with recombinant DNA technology has paved the way for tailor-made and targeted medicines. Gene- and cell-based therapies are emerging as new approaches.
Recombinant therapeutic proteins are of a complex nature (composed of a long chain of amino acids, modified amino acids, derivatized by sugar moieties, folded by complex mechanisms). These proteins are made in living cells (bacteria, yeast, animal or human cell lines). The ultimate characteristics of a drug containing a recombinant therapeutic protein are to a large part determined by the process through which they are produced: choice of the cell type, development of the genetically modified cell for production, production process, purification process, formulation of the therapeutic protein into a drug.
After the expiry of the patent of approved recombinant drugs (e.g., insulin, human growth hormone, interferons, erythropoietin, monoclonal antibodies and more) any other biotech company can develop and market these biologics (thus called biosimilars).
The typical reference product has undergone numerous changes in its manufacturing processes, and such changes in the manufacturing process (ranging from a change in the supplier of cell culture media to new purification methods or new manufacturing sites) was substantiated with appropriate data and was approved by the EMA.
The current concept of development of biosimilar mAbs follows the principle that an extensive state of the art physicochemical, analytical and functional comparison of the molecules is complemented by comparative non-clinical and clinical data that establish equivalent efficacy and safety in a clinical "model" indication that is most sensitive to detect any minor differences (if these exist) between the biosimilar and its reference mAb also at the clinical level.
The EMA has recognized this fact, which has resulted in the establishment of the term "biosimilar" in recognition that, whilst biosimilar products are similar to the original product, they are not exactly the same.
Originally the complexity of biological molecules led to requests for substantial efficacy and safety data for a biosimilar approval. This has been progressively replaced with a greater dependence on assays, from quality through to clinical, that show assay sensitivity sufficient to detect any significant difference in dose. However, the safe application of biologics depends on an informed and appropriate use by healthcare professionals and patients. Introduction of biosimilars also requires a specifically designed pharmacovigilance plan. It is difficult and costly to recreate biologics because the complex proteins are derived from living organisms that are genetically modified. In contrast, small molecule drugs made up of a chemically based compound can be easily replicated and are considerably less expensive to reproduce. In order to be released to the public, biosimilars must be shown to be as close to identical to the parent innovator biologic product based on data compiled through clinical, animal, analytical studies and conformational status.
Generally, once a drug is released in the market by the FDA, it has to be re-evaluated for its safety and efficacy once every six months for the first and second years. Afterward, re-evaluations are conducted yearly, and the result of the assessment should be reported to authorities such as FDA. Biosimilars are required to undergo pharmacovigilance (PVG) regulations as its reference product. Thus biosimilars approved by the EMA are required to submit a risk management plan (RMP) along with the marketing application and have to provide regular safety update reports after the product is in the market. The RMP includes the safety profile of the drug and proposes the prospective pharmacovigilance studies.
Several PK studies, such as studies conducted by Committee for Medicinal Products for Human Use (CHMP), have been conducted under various ranges of conditions; Antibodies from an originator's product versus antibodies from a biosimilar; combination therapy and monotherapy; various diseases, etc. on the purpose to verify comparability in pharmacokinetics of the biosimilar with the reference medicinal product in a sufficiently sensitive and homogeneous population.
Nomenclature
In the European Union, no unique identifier of a biosimilar medicine product is required, as the same rules are followed as for all biologics.
The US decided on a different approach, requiring the assignment of a four-letter suffix to the nonproprietary name of the original product to distinguish between innovator drugs and their biosimilars. Japan has similar requirements. The suffix approach has been criticized on the grounds of compromising the INN system and delaying the marketing of biosimilars. Australia decided not to use a 4-letter suffix.
A version of the four-letter suffix has been proposed to the WHO as the biological qualifier (BQ). It is not part of the international nonproprietary name (INN), but is proposed to be managed under the same registry. The report 1 of the May 2017 WHO Expert Consultation on Improving Access to and Use of Similar Biotherapeutic Products, published in October 2017, revealed on page 4, that following the outcome arising from the meeting: "No consensus was reached on whether WHO should continue with the BQ… WHO will not be proceeding with this at present."
Australia
Biosimilars available in Australia include adalimumab, bevacizumab, enoxaparin, epoetin lambda, etanercept, filgrastim, follitropin alfa, infliximab, insulin aspart, insulin glargine, pegfilgrastim, rituximab, teriparatide, and trastuzumab.
European Union
Biosimilar medicines approved in the European Union (EU) are interchangeable with their reference medicine or with an equivalent biosimilar.
| Active substance | Reference product | Biosimilar medicines |
|---|---|---|
| Adalimumab | Humira | Amgevita, Amsparity, Cyltezo, Halimatoz, Hefiya, Hukyndra, Hulio, Hyrimoz, Idacio, Imraldi, Kromeya, Libmyris, Solymbic, Trudexa, Yuflyma |
| Bevacizumab | Avastin | Abevmy, Alymsys, Aybintio, Equidacent, Mvasi, Onbevzi, Oyavas, Zirabev |
| Enoxaparin sodium | Lovenox | Inhixa |
| Epoetin alfa | Eprex/Erypo | Abseamed, Binocrit, Epoetin Alfa Hexal, Retacrit, Silapo |
| Etanercept | Enbrel | Benepali, Erelzi, Nepexto |
| Filgrastim | Neupogen | Accofil, Filgrastim Hexal, Grastofil, Nivestim, Ratiograstim, Tevagrastim, Zarzio |
| Follitropin alfa | Gonal-F | Bemfola, Ovaleap |
| Infliximab | Remicade | Flixabi, Inflectra, Remsima, Zessly |
| Insulin aspart | NovoRapid | Fiasp, Insulin aspart Sanofi, Kirsty, NovoMix, Ryzodeg |
| Insulin glargine | Lantus | Abasaglar, Semglee |
| Insulin lispro | Humalog | Insulin lispro Sanofi |
| Pegfilgrastim | Neulasta | Cegfila, Fulphila, Grasustek, Pelgraz, Pelmeg, Udenyca, Stimufend, Ziextenzo |
| Ranibizumab | Lucentis | Byooviz, Ranivisio, Ximluci |
| Rituximab | MabThera | Blitzima, Ritemvia, Rituzena, Rixathon, Riximyo, Ruxience, Truxima |
| Somatropin | Genotropin | Omnitrope |
| Teriparatide | Forsteo | Movymia, Terrosa |
| Trastuzumab | Herceptin | Herzuma, Kanjinti, Ogivri, Ontruzant, Trazimera, Zercepac |
United States
BPCI Act
The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) was originally sponsored and introduced on June 26, 2007, by Senator Edward Kennedy (D-MA). It was formally passed under the Patient Protection and Affordable Care Act (PPAC Act), signed into law by President Barack Obama on March 23, 2010. The BPCI Act was an amendment to the Public Health Service Act (PHS Act) to create an abbreviated approval pathway for biological products that are demonstrated to be highly similar (biosimilar) to a Food and Drug Administration (FDA) approved biological product. The BPCI Act is similar, conceptually, to the Drug Price Competition and Patent Term Restoration Act of 1984 (also referred to as the "Hatch-Waxman Act") which created biological drug approval through the Federal Food, Drug, and Cosmetic Act (FFD&C Act). The BPCI Act aligns with the FDA's longstanding policy of permitting appropriate reliance on what is already known about a drug, thereby saving time and resources and avoiding unnecessary duplication of human or animal testing. The FDA has released a total of four draft guidelines related to biosimilar or follow-on biologics development. Upon the release of the first three guidance documents the FDA held a public hearing on May 11, 2012.
In 2018, the FDA released a Biosimilars Action Plan to implement regulations from the BPCI, including limiting the abuse of the Risk Evaluation and Mitigation Strategy (REMS) system for evergreening and transitioning insulin and human growth hormone to regulation as biologics rather than drugs.
US approved biosimilars
| Date of Biosimilar FDA Approval | Biosimilar Product | Original Product |
|---|---|---|
| March 6, 2015 | filgrastim-sndz/Zarxio | filgrastim/Neupogen |
| April 5, 2016 | infliximab-dyyb/Inflectra | infliximab/Remicade |
| August 30, 2016 | etanercept-szzs/Erelzi | etanercept/Enbrel |
| September 23, 2016 | adalimumab-atto/Amjevita | adalimumab/Humira |
| April 21, 2017 | infliximab-abda/Renflexis | infliximab/Remicade |
| August 25, 2017 | adalimumab-adbm/Cyltezo | adalimumab/Humira |
| September 14, 2017 | bevacizumab-awwb/Mvasi | bevacizumab/Avastin |
| December 1, 2017 | trastuzumab-dkst/Ogivri | trastuzumab/Herceptin |
| December 13, 2017 | infliximab-qbtx/Ixifi | infliximab/Remicade |
| May 15, 2018 | epoetin alfa-epbx/Retacrit | epoetin alfa/Procrit |
| June 4, 2018 | pegfilgrastim-jmdb/Fulphila | pegfilgrastim/Neulasta |
| July 20, 2018 | filgrastim-aafi/Nivestym | filgrastim/Neupogen |
| October 30, 2018 | adalimumab-adaz/Hyrimoz | adalimumab/Humira |
| November 2, 2018 | pegfilgrastim-cbqv/Udenyca | pegfilgrastim/Neulasta |
| November 28, 2018 | rituximab-abbs/Truxima | rituximab/Rituxan |
| December 14, 2018 | trastuzumab-pkrb/Herzuma | trastuzumab/Herceptin |
| January 18, 2019 | trastuzumab-dttb/Ontruzant | trastuzumab/Herceptin |
| March 11, 2019 | trastuzumab-qyyp/Trazimera | trastuzumab/Herceptin |
| April 25, 2019 | etanercept-ykro/Eticovo | etanercept/Enbrel |
| June 13, 2019 | trastuzumab-anns/Kanjinti | trastuzumab/Herceptin |
| June 27, 2019 | bevacizumab-bvzr/Zirabev | bevacizumab/Avastin |
| July 23, 2019 | rituximab-pvvr/Ruxience | rituximab/Rituxan |
| July 23, 2019 | adalimumab-bwwd/Hadlima | adalimumab/Humira |
| November 4, 2019 | pegfilgrastim-bmez/Ziextenzo | pegfilgrastim/Neulasta |
| November 15, 2019 | adalimumab-afzb/Abrilada | adalimumab/Humira |
| December 6, 2019 | infliximab-axxq/Avsola | infliximab/Remicade |
| June 10, 2020 | pegfilgrastim-apgf/Nyvepria | pegfilgrastim/Neulasta |
| July 6, 2020 | adalimumab-fkjp/Hulio | adalimumab/Humira |
| December 17, 2020 | rituximab-arrx/Riabni | rituximab/Rituxan |
| July 28, 2021 | insulin glargine-yfgn/Semglee | insulin glargine/Lantus |
| September 17, 2021 | ranibizumab-nuna/Byooviz | ranibizumab/Lucentis |
| December 17, 2021 | insulin glargine-aglr/Rezvoglar | insulin glargine/Lantus |
| December 17, 2021 | adalimumab-aqvh/Yusimry | adalimumab/Humira |
| February 25, 2022 | filgrastim-ayow/Releuko | filgrastim/Neupogen |
| April 13, 2022 | bevacizumab-maly/Alymsys | bevacizumab/Avastin |
| May 26, 2022 | pegfilgrastim-pbbk/Fylnetra | pegfilgrastim/Neulasta |
| August 2, 2022 | ranibizumab-eqrn/Cimerli | ranibizumab/Lucentis |
| September 1, 2022 | pegfilgrastim-fpgk/Stimufend | pegfilgrastim/Neulasta |
| September 27, 2022 | bevacizumab-adcd/Vegzelma | bevacizumab/Avastin |
| December 13, 2022 | adalimumab-aacf/Idacio | adalimumab/Humira |
Proposed reforms
In the United States, biosimilars have not had the expected impact on prices, leading a 2019 proposal to price regulate instead after an exclusivity period. Another proposal requires originators to share the underlying cell lines.
In 2019, the proposed Biologic Patent Transparency Act would help address evergreening "patent thickets" by requiring that all patents protecting a biosimilar be disclosed.
Biosimilars have found it difficult to get market share, which led biosimilar developer Pfizer to sue Johnson & Johnson over anticompetitive contracts with pharmacy benefit managers which bundle discounts; these are sometimes called the "rebate wall", and the rebates are generally unavailable to customers.
Market implications
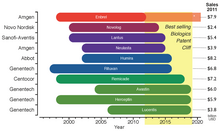
The legal requirements of approval pathways, together with the costly manufacturing processes, escalates the developing costs for biosimilars that could be between $75–$250 million per molecule. This market entry barrier affects not only the companies willing to produce them but could also delay availability of inexpensive alternatives for public healthcare institutions that subsidize treatment for their patients. Even though the biosimilars market is rising, the price drop for biological drugs at risk of patent expiration will not be as great as for other generic drugs; in fact it has been estimated that the price for biosimilar products will be 65%-85% of their originators. Biosimilars are drawing market's attention since there is an upcoming patent cliff, which will put nearly 36% of the $140 billion market for biologic drugs at risk (as of 2011), this considering only the top 10 selling products.
The global biosimilars market was US$1.3 billion in 2013, and is expected to reach US$35 billion by 2020, driven by the patent expiration of additional ten blockbuster biologic drugs.
Companies
Certain companies (in some cases subsidiaries) tend to operate as generic drug manufacturers, with major ones including Teva, Viatris, and Sandoz and may also extend that focus to biosimilars. Sandoz, for example, introduced the first biosimilar in the United States. By value the most important biosimilar business is run by Celltrion (South Korea). Newer companies such as India-based Cadila Pharmaceuticals, Sun Pharma, Aurobindo Pharma, Lupin and Dr. Reddy's Laboratories as well as Canada-based Apotex have taken share in traditional generics, which has led older companies to shift their focus to complex drugs such as biosimilars.
Further reading
- Udpa N, Million RP (January 2016). "Monoclonal antibody biosimilars". Nature Reviews. Drug Discovery. 15 (1): 13–4. doi:10.1038/nrd.2015.12. PMID 26678619. S2CID 27954836.
- Jelkmann W (October 2010). "Biosimilar epoetins and other "follow-on" biologics: update on the European experiences". American Journal of Hematology. 85 (10): 771–80. doi:10.1002/ajh.21805. PMID 20706990. S2CID 205293428.
- "New guide on biosimilar medicines for healthcare professionals". Prepared Jointly by the European Medicines Agency and the European Commission. Retrieved May 10, 2017.
- "What's keeping less expensive biologic drugs from the U.S. market?". PBS NewsHour. April 19, 2014.
-
Therapeutics, Initiative (November 2019). "Biosimilars or Biologics: What's the difference?". Therapeutics Letter 123.
