
The discovery of an orally inactive peptide from snake venom established the unimportant role of angiotensin converting enzyme (ACE) inhibitors in regulating blood pressure. This led to the development of captopril, the first ACE inhibitor. When the adverse effects of captopril became apparent new derivates were designed. Then after the discovery of two active sites of ACE: N-domain and C-domain, the development of domain-specific ACE inhibitors began.
Development of first generation ACE inhibitors
The development of the nonapeptide teprotide (Glu-Trp-Pro-Arg-Pro-Gln-Ile-Pro-Pro), which was originally isolated from the venom of the Brazilian pit viper Bothrops jararaca, greatly clarified the importance of ACE in hypertension. However, its lack of oral activity limited its therapeutic utility.
L-benzylsuccinic acid (2(R)-benzyl-3-carboxypropionic acid) was described to be the most potent inhibitor of carboxypeptidase A in the early 1980s. The authors referred to it as a by-product analog and it was proposed to bind to the active site of carboxypeptidase A via succinyl carboxyl group and a carbonyl group. Their findings established that L-benzylsuccinic acid is bound at a single locus at the active site of carboxypeptidase A. The authors discussed but dismissed the suggestion that the carboxylate function might bind to the catalytically functional zinc ion present at the active site. Later however this was found to be the case.
Drug design of captopril (sulfhydrils)
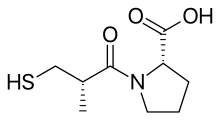
Over 2000 compounds were tested randomly in a guinea pig ileum test and succinyl-L-proline was found to have the properties of a specific ACE inhibitor. It showed inhibitory effect of angiotensin I and bradykinin without having any effects on angiotensin II. Then researchers started to search for a model that would explain inhibition on the basis of specific interactions of compounds with the active site of ACE. Previous studies with substrates and inhibitors of ACE suggested that it was a zinc-containing metalloprotein and a carboxypeptidase similar to pancreatic carboxypeptidase A. However ACE releases dipeptides rather than single amino acids from the C-terminus of the peptide substrates. And it was assumed that both their mechanism of action and their active site might be similar. A positively charged Arg145 at the active site was thought to bind with the negatively charged C-terminal carboxyl group of the peptide substrate. It was also proposed that ACE binds by hydrogen bonding to the terminal, non scissile, peptide bond of the substrate.
But since ACE is a dipeptide carboxypeptidase, unlike carboxypeptidase A, the distance between the cationic carboxyl-binding site and the zinc atom should be greater, by approximately the length of one amino acid residue. Proline was chosen as the amino acid moiety because of its presence as the carboxy terminal amino acid residue in teprotide and other ACE inhibitors found in snake venoms. 11 other amino acids were tested but none of them were more inhibitory. So it was proposed that succinyl amino acid derivative should be an ACE inhibitor and succinyl-L-proline was found to be such an inhibitor.
It was also known that the nature of penultimate amino acid residue of a peptide substrate for ACE influences binding to the enzyme. The acyl group of the carboxyalkanoyl amino acid binds the zinc ion of the enzyme and occupies the same position at the active site of ACE as the penultimate. Therefore, the substituent of the acyl group might also influence binding to the enzyme. A 2-methyl substituent with D configuration was found to enhance the inhibitory potency by about 15 fold of succinyl-L-proline. Then the search for a better zinc-binding group started. Replacement of the succinyl carboxyl group by nitrogen-containing functionalities (amine, amide or guanidine) did not enhance inhibitory activity. However a potency breakthrough was achieved by the replacement of the carboxyl group with a sulfhydryl function (SH), a group with greater affinity for the enzyme bound zinc ion. This yielded a potent inhibitor that was 1000 times more potent than succinyl-L-proline. The optimal acyl chain length for mercaptoalkanoyl derivates of proline was found to be 3-mercaptopropanoyl-L-proline, 5 times greater than that of 2-mercaptoalkanoyl derivates and 50 times greater than that of 4-mercaptoalkanoyl derivates. So the D-3-mercapto-2-methylpropanoyl-L-proline or Captopril was the most potent inhibitor. Later, the researchers compared a few mercaptoacyl amino acid inhibitors and concluded that the binding of the inhibitor to the enzyme involved a hydrogen bond between a donor site on the enzyme and the oxygen of the amide carbonyl, much like predicted for the substrates.
Drug design of other first generation ACE inhibitors


The most common adverse effects of Captopril, skin rash and loss of taste, are the same as caused by mercapto-containing penicillamine. Therefore, a group of researchers aimed at finding potent, selective ACE inhibitors that would not contain a mercapto (SH) function and would have a weaker chelating function. They returned to work with carboxyl compounds and started working with substituted N-carboxymethyl-dipeptides as a general structure (R-CHCOOH-A1-A2). According to previous research they assumed that cyclic imino acids would result in good potency if substituted on the carboxyl terminus of the dipeptide. Therefore, substituting A2 with proline gave good results. They also noted that according to the enzyme's specificity imino acids in the position next to the carboxyl terminus would not give a potent compound. By substituting R and A1 groups with hydrophobic and basic residues would give a potent compound. By substituting –NH in the general structure resulted in loss of potency which is consistent to the enzyme's need for a –NH in corresponding position on the substrates. The results were 2 active inhibitors: Enalaprilat and Lisinopril. These compounds both have phenylalanine in R position which occupies the S1 groove in the enzyme. The result was thus these two new, potent tripeptide analogues with zinc-coordinating carboxyl group: Enalaprilat and Lisinopril.
Discovery of 2 active sites: C-domain and N-domain
Most of the ACE inhibitors on the market today are non-selective towards the two active sites of ACE because their binding to the enzyme is based mostly on the strong interaction between the zinc atom in the enzyme and the strong chelating group on the inhibitor. The resolution of the 3D structure of germinal ACE, which has only one active site that corresponds with C-domain of the somatic ACE, offers a structural framework for structure-based design approach. Although N- and C-domain have comparable rates in vitro of ACE hydrolyzing, it seems like that in vivo the C-domain is mainly responsible for regulating blood pressure. This indicates that C-domain selective inhibitors could have similar profile to that of a current non-selective inhibitors. Angiotensin I is mainly hydrolyzed by the C-domain in vivo but bradykinin is hydrolyzed by both active sites. Thus, by developing a C-domain selective inhibitor would permit some degradation of bradykinin by the N-domain and this degradation could be enough to prevent accumulation of excess bradykinin which has been observed during attacks of angioedema. C-domain selective inhibition could possibly result in specialized control of blood pressure with less vasodilator-related adverse effects. N-domain selective inhibitors on the other hand give the possibility of opening up novel therapeutic areas. Apparently, the N-domain does not have a big role in controlling blood pressure but it seems to be the principal metabolizing enzyme for AcSDKP, a natural haemoregulatory hormone.
Drug design of Keto-ACE and its ketomethylene derivatives
It was found that other carbonyl-containing groups such as ketones could substitute for the amide bond that links Phe and Gly in ACE inhibitors. Keto-ACE, first described in 1980, has emerged as a potential lead compound for C-domain specific ACE inhibitors. Keto-ACE, a tripeptide analogue of Phe-Gly-Pro, contains a bulky P1 and P2benzyl ring and was shown to inhibit the hydrolysis of angiotensin I and bradykinin via the C-domain. The synthesis of keto-ACE analogues with Trp or Phe at the P2’ position led to a marked increase in C-domain selectivity, but the introduction of an aliphatic P2 group conferred N-domain selectivity. Inhibitory potency may further be enhanced by the incorporation of hydrophobic substituent, such as phenyl group at the P1’ position. P1’ substituents with S-stereochemistry have also been shown to possess greater inhibitory potency than their R-counterparts.
Keto-ACE was used as the basis for the design of ketomethylene derivates. Its analogues contain a ketomethylene isostere replacement at the scissile bond that is believed to mimic the tetrahedron transition state of the proteolytic reaction at the active site. The focus was on a simple tripeptide Phe-Ala-Pro, which in earlier enzyme assays has shown inhibition activity. Replacement of alanine with glycin gave a tripeptide with 1/14th of the inhibition activity of Phe-Ala-Pro. The benzoylated derivative of Phe-Gly-Pro, Bz-Phe-Gly-Pro, was twice as active. To reduce the peptidic nature of ketomethylene inhibitors the P1’ and P2’ substituent may be cyclized to form a lactam, where there is a correlation between the inhibitory potency and the ring size. In 2001 it was postulated that a substitution α to nitrogen and making of 3-methyl-substituted analog of A58365A, a pyridone acid isolated from the fermentation broth of the bacterium Streptomyces chromofuscus with ACE inhibitory activity, might influence the level of biological activity by steric or hydrophobic effect, and/or by preventing reactions at C3. It was also noticed during the synthetic work on A58365A that potential precursors were sensitive to oxidation of the five-membered ring and so the 3-methyl analogue might be more stable in this respect.
Drug design of silanediol
The fact that carbon and silicone have similar, but also dissimilar, characteristics triggered the interest in substituting carbon with silanediol as a central, zinc chelating group. Silicone forms a dialkylsilanediol compound that is sufficiently hindered so the formation of a siloxane polymer does not occur. Silanediols are more stable than carbon diols so they are expected to have longer half-life. Silanediols are also neutral at physiological pH (do not ionize). Four stereoisomers of Phe-Ala silanediol were compared to ketone-based inhibitors and the silanediol were found to be fourfold less potent than the ketone analogue. This is because silanediols are weaker zinc chelators compared with ketones. Replacement of the silanediol, with a methylsilano group gave little enzyme inhibition. This confirms that the silanediol group interacts with ACE as a transition state analogue and the interaction is in a manner similar to that of ketone. If the benzyl group of silanediol is replaced by an i-butyl group it gives a weaker ACE inhibitor. Introduction of a hydrophobic methyl phenyl gives a little more potency than an analogue with a tert-butyl-group at P1. That suggests that methyl phenyl gives a better S1 recognition than a tert-butyl group.
Phosphinic peptides
Phosphinic peptides are pseudo-peptides where a phosphinic acid bond (PO2-CH-) has replaced a peptide bond in the peptide analogue sequence. To some extent the chemical structure of phosphinic peptides is similar to that of intermediates which are produced in hydrolysis of peptides by proteolytic enzymes. The hypothesis has been made that these pseudo-peptides mimic the structure of the enzyme substrates in their transition state and crystallography of zinc proteases in complex with phosphinic peptides supports that hypothesis.
Drug design of RXP 407
RXP 407 is the first N-domain selective phosphinic peptide and was discovered by screening phosphinic peptides libraries. Before the discovery of RXP 407 it had long been claimed that the free C-terminal carboxylate group in P2’ position was essential to the potency of ACE inhibitor so it can be reasoned that this has postponed the discovery of N-domain selective ACE inhibitors. When RXP 407 was discovered researchers looked into phosphinic peptides with 3 different general formula, each containing 2 unidentified amino acids, only 1 of these general formula showed potent inhibition (Ac-Yaa-Pheψ(PO2-CH2)Ala-Yaa’-NH2). Peptide mixtures were made, substituting Yaa and Yaa’ with different amino acids, trying to establish if there would be a potent inhibitor that could inhibit either the N-domain or the C-domain of the enzyme. The result was that the compound Ac-Asp(L)-Pheψ(PO2-CH2)(L)Ala-Ala-NH2 actively inhibited the N-domain and was given the name RXP 407. Structure-function relationship showed that the C-terminus carboxamide group played a crucial role in the selectivity for the N-domain of ACE. Additionally, the N-acetyl group and the aspartic side chain in the P2 position aides in the N-domain selectivity of the inhibitor. These features make the inhibitor inaccessible to the C-domain but give good potency for the N-domain, this leads to a difference in inhibitory potency of the active sites of three orders of magnitude. These results also indicate that the N-domain possess a broader selectivity than the C-domain. Another difference between the older ACE inhibitors and RXP 407 is the molecular size of the compound. The older ACE inhibitors had mostly been interacting with S1’, S2’ and S1 subsites but RXP 407 interacts in addition with the S2 subsite. This also is important for the selectivity of the inhibitor since the aspartic side chain and N-acetyl group are located in the P2 position.
Drug design of RXPA 380
RXPA380 was the first inhibitor that was highly selective of the C-domain of ACE, it has the formula Phe-Phe-Pro-Trp. The development of this compound was built on researches that showed that some bradykinin-potentiating peptides showed selectivity for the C-domain and all had several prolines in their structure. These observations lead the researchers to synthesize phosphinic peptides containing a proline residue in the P1’ position and evaluating these compounds led to the discovery of RXPA380. To study the roles of the residues on RXPA380 the researchers made 7 analogues of RXPA380. All of the compounds made were obtained as a mixture of either 2 or 4 diastereoisomers but all of them were easily resolved and only one of them was potent. This is consistent with the initial modeling studies of RXPA380 which showed that only one diastereomer could accommodate in the active site of germinal ACE. Analogues where pseudo-proline or tryptophan residues had been substituted showed less selectivity than RXPA380. This is probably because these two analogues have more potency toward the N-domain than RXPA380 does. Substituting both of these residues gives great potency but none selectivity. This shows that pseudo-proline and tryptophan residues accommodate well in the C-domain but not in the N-domain. 2 more analogues with both pseudo-proline and tryptophan but missing the pseudo-phenylalanine residue in P1 position showed low potency for N-domain, similar to RXPA380. This supports the significant role of these two residues in the selectivity for C-domain. These two analogues also have less potency for the C-domain which shows that the C-domain prefers pseudo-phenylalanine group in P1 position. Modeling of RXPA380-ACE complex showed that the pseudo-proline residue of the inhibitor was surrounded by amino acids similar to that of the N-domain thus interactions with S2’ domain might not be responsible for the selectivity of RXPA380. 7 of 12 amino acids surrounding tryptophan are the same in C- and N-domain, the biggest difference is that 2 bulky and hydrophobic amino acids in the C-domain have been replaced with 2 smaller and polar amino acids in the N-domain. This indicates that low potency of RXPA380 for N-domain is not because the S2’ cavity does not accommodate the tryptophan side chain but rather that important interactions are missing between the tryptophan side chain and the amino acids of the C-domain. Based on the proximity between the tryptophan side chain and Asp1029 there is also a possible hydrogen bond between the carboxylate of Asp1029 and the NH indole ring in the C-domain but this interaction is much weaker in the N-domain.