
| Fraser syndrome | |
|---|---|
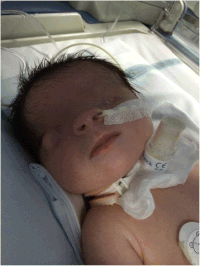 | |
| Bilateral cryptophthalmos with microphthalmos in the left ocular globe and abnormal right ocular globe in a female infant with Fraser syndrome. | |
| Specialty |
Medical genetics |
Fraser syndrome (also known as Meyer-Schwickerath's syndrome, Fraser-François syndrome, or Ullrich-Feichtiger syndrome) is an autosomal recessive congenital disorder, identified by several developmental anomalies. Fraser syndrome is named for the geneticist George R. Fraser, who first described the syndrome in 1962.
Signs and symptoms

It is characterized by developmental defects including cryptophthalmos (where the eyelids fail to separate in each eye), and intersex development in the genitals (such as micropenis, or clitoromegaly) and cryptorchidismCongenital malformations of the nose, ears, larynx and renal system, as well as developmental delays, manifest occasionally.Syndactyly (fused fingers or toes) has also been noted.
Genetics

The genetic background of this disease has been linked to a gene called FRAS1, which seems to be involved in skin epithelial morphogenesis during early development. It has also been associated with FREM2 and with GRIP1.
Mapping
By autozygosity mapping, McGregor et al. (2003) located the Fraser syndrome locus to chromosome 4q21.
Genetic Heterogeneity
In 6 of 18 consanguineous families with Fraser syndrome, van Haelst et al. (2008) excluded linkage to both the FRAS1 and FREM2 genes, indicating genetic heterogeneity.
Molecular genetics
In 5 families with Fraser syndrome, McGregor et al. (2003) identified 5 homozygous mutations in the FRAS1 gene (e.g., 607830.0001), which encodes a putative extracellular matrix (ECM) protein.
In 2 families with Fraser syndrome unlinked to the FRAS1 gene, Jadeja et al. (2005) found a homozygous missense mutation in the FREM2 gene (608945.0001).
In an infant girl with Fraser syndrome, Slavotinek et al. (2006) identified compound heterozygosity for a deletion (607830.0006) and an insertion (607830.0007) in the FRAS1 gene, inherited from her mother and her father, respectively.
Cavalcanti et al. (2007) described 2 stillborn Brazilian male siblings, born at 25 and 29 weeks' gestation, respectively. One sibling appeared to have a lethal form of ablepharon-macrostomia syndrome (AMS; 200110) or an intermediate phenotype between AMS and Fraser syndrome, and the other had classic Fraser syndrome. Analysis of the FRAS1 gene revealed homozygosity for a splice site mutation (607830.0008), resulting in a severely truncated protein in both siblings and heterozygosity for the mutation in both parents. Cavalcanti et al. (2007) concluded that a phenotype resembling AMS is a rare clinical expression of Fraser syndrome, with no obvious genotype/phenotype correlation.
In a female fetus with a normal karyotype and cryptophthalmos, ambiguous external genitalia, syndactyly, bilobed lungs, bilateral renal agenesis, hypoplastic bladder, and agenesis of internal genitalia with streak ovaries, Shafeghati et al. (2008) identified homozygosity for a splice site mutation in the FREM2 gene (608945.0002). The consanguineous Iranian parents were heterozygous for the mutation. An earlier pregnancy had resulted in the intrauterine death at 30 weeks of gestation of a male fetus with a normal karyotype in whom the diagnosis of Fraser syndrome was suggested by the presence of cryptophthalmos, syndactyly, ambiguous genitalia, imperforate anus, bilateral renal agenesis, pulmonary hypoplasia, and hydrocephalus. The authors noted that the findings in the sibs were consistent with classic Fraser syndrome.
Among 18 consanguineous families with Fraser syndrome, van Haelst et al. (2008) found 9 families with linkage to FRAS1, 3 families to FREM2, and 3 families to both genes. Six families did not link to either locus, indicating genetic heterogeneity. Among a larger group of 33 families, including the 18 consanguineous families, molecular analysis identified 11 novel mutations in the FRAS1 gene in 10 families and 1 mutation in the FREM2 gene (608945.0003) in 1 family. A literature review of genotype/phenotype correlations suggested that patients with FRAS1 mutations have more frequent skull ossification defects and a low insertion of the umbilical cord compared to patients without a FRAS1 mutation, but the findings were not statistically significant.
Diagnosis

The diagnosis of this syndrome can be made on clinical examination and perinatal autopsy.
Koenig and Spranger (1986) noted that eye lesions are apparently nonobligatory components of the syndrome. The diagnosis of Fraser syndrome should be entertained in patients with a combination of acrofacial and urogenital malformations with or without cryptophthalmos. Thomas et al. (1986) also emphasized the occurrence of the cryptophthalmos syndrome without cryptophthalmos and proposed diagnostic criteria for Fraser syndrome. Major criteria consisted of cryptophthalmos, syndactyly, abnormal genitalia, and positive family history. Minor criteria were congenital malformation of the nose, ears, or larynx, cleft lip and/or palate, skeletal defects, umbilical hernia, renal agenesis, and intellectual disability. Diagnosis was based on the presence of at least 2 major and 1 minor criteria, or 1 major and 4 minor criteria.
Boyd et al. (1988) suggested that prenatal diagnosis by ultrasound examination of eyes, digits, and kidneys should detect the severe form of the syndrome. Serville et al. (1989) demonstrated the feasibility of ultrasonographic diagnosis of the Fraser syndrome at 18 weeks' gestation. They suggested that the diagnosis could be made if 2 of the following signs are present: obstructive uropathy, microphthalmia, syndactyly, and oligohydramnios. Schauer et al. (1990) made the diagnosis at 18.5 weeks' gestation on the basis of sonography. Both the female fetus and the phenotypically normal father had a chromosome anomaly: inv(9)(p11q21). An earlier born infant had Fraser syndrome and the same chromosome 9 inversion.
Van Haelst et al. (2007) provided a revision of the diagnostic criteria for Fraser syndrome according to Thomas et al. (1986) through the addition of airway tract and urinary tract anomalies to the major criteria and removal of intellectual disability and clefting as criteria. Major criteria included syndactyly, cryptophthalmos spectrum, urinary tract abnormalities, ambiguous genitalia, laryngeal and tracheal anomalies, and positive family history. Minor criteria included anorectal defects, dysplastic ears, skull ossification defects, umbilical abnormalities, and nasal anomalies. Cleft lip and/or palate, cardiac malformations, musculoskeletal anomalies, and intellectual disability were considered uncommon. Van Haelst et al. (2007) suggested that the diagnosis of Fraser syndrome can be made if either 3 major criteria, or 2 major and 2 minor criteria, or 1 major and 3 minor criteria are present in a patient.
Epidemiology
The incidence of Fraser syndrome is 0.043 per 10,000 live born infants and 1.1 in 10,000 stillbirths, making it a rare syndrome.