
A glycosidic bond or glycosidic linkage is a type of ether bond that joins a carbohydrate (sugar) molecule to another group, which may or may not be another carbohydrate.
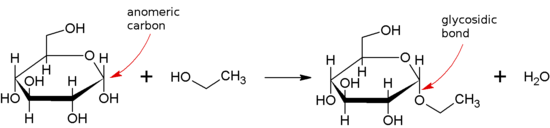
A glycosidic bond is formed between the hemiacetal or hemiketal group of a saccharide (or a molecule derived from a saccharide) and the hydroxyl group of some compound such as an alcohol. A substance containing a glycosidic bond is a glycoside.
The term 'glycoside' is now extended to also cover compounds with bonds formed between hemiacetal (or hemiketal) groups of sugars and several chemical groups other than hydroxyls, such as -SR (thioglycosides), -SeR (selenoglycosides), -NR1R2 (N-glycosides), or even -CR1R2R3 (C-glycosides).
Particularly in naturally occurring glycosides, the compound ROH from which the carbohydrate residue has been removed is often termed the aglycone, and the carbohydrate residue itself is sometimes referred to as the 'glycone'.
S-, N-, C-, and O-glycosidic bonds
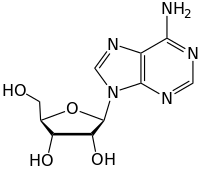
Glycosidic bonds of the form discussed above are known as O-glycosidic bonds, in reference to the glycosidic oxygen that links the glycoside to the aglycone or reducing end sugar. In analogy, one also considers S-glycosidic bonds (which form thioglycosides), where the oxygen of the glycosidic bond is replaced with a sulfur atom. In the same way, N-glycosidic bonds, have the glycosidic bond oxygen replaced with nitrogen. Substances containing N-glycosidic bonds are also known as glycosylamines. C-glycosyl bonds have the glycosidic oxygen replaced by a carbon; the term "C-glycoside" is considered a misnomer by IUPAC and is discouraged. All of these modified glycosidic bonds have different susceptibility to hydrolysis, and in the case of C-glycosyl structures, they are typically more resistant to hydrolysis.
Numbering, and α/β distinction of glycosidic bonds

When an anomeric center is involved in a glycosidic bond (as is common in nature) then one can distinguish between α- and β-glycosidic bonds by the relative stereochemistry of the anomeric position and the stereocenter furthest from C1 in the saccharide.
Pharmacologists often join substances to glucuronic acid via glycosidic bonds in order to increase their water solubility; this is known as glucuronidation. Many other glycosides have important physiological functions.
Chemical approaches
Nüchter et al. (2001) have shown a new approach to Fischer glycosidation. Employing a microwave oven equipped with refluxing apparatus in a rotor reactor with pressure bombs, Nüchter et al. (2001) were able to achieve 100% yield of α- and β-D-glucosides. This method can be performed on a multi-kilogram scale.
- Vishal Y Joshi's method
Joshi et al. (2006) propose the Koenigs-Knorr method in the stereoselective synthesis of alkyl D-glucopyranosides via glycosylation, with the exception of using lithium carbonate which is less expensive and toxic than the conventional method of using silver or mercury salts. D-glucose is first protected by forming the peracetate by addition of acetic anhydride in acetic acid, and then addition of hydrogen bromide which brominates at the 5-position. On addition of the alcohol ROH and lithium carbonate, the OR replaces the bromine and on deprotecting the acetylated hydroxyls the product is synthesized in relatively high purity. It was suggested by Joshi et al. (2001) that lithium acts as the nucleophile that attacks the carbon at the 5-position and through a transition state the alcohol is substituted for the bromine group. Advantages of this method as well as its stereoselectivity and low cost of the lithium salt include that it can be done at room temperature and its yield compares relatively well with the conventional Koenigs-Knorr method.
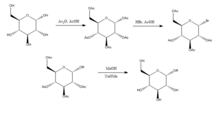
Glycoside hydrolases
Glycoside hydrolases (or glycosidases), are enzymes that break glycosidic bonds. Glycoside hydrolases typically can act either on α- or on β-glycosidic bonds, but not on both. This specificity allows researchers to obtain glycosides in high epimeric excess, one example being Wen-Ya Lu's conversion of D-Glucose to Ethyl β-D-glucopyranoside using naturally-derived glucosidase. It is worth noting that Wen-Ya Lu utilized glucosidase in a reverse manner opposite to the enzyme's biological functionality:

Glycosyltransferases
Before monosaccharide units are incorporated into glycoproteins, polysaccharides, or lipids in living organisms, they are typically first "activated" by being joined via a glycosidic bond to the phosphate group of a nucleotide such as uridine diphosphate (UDP), guanosine diphosphate (GDP), thymidine diphosphate (TDP), or cytidine monophosphate (CMP). These activated biochemical intermediates are known as sugar nucleotides or sugar donors. Many biosynthetic pathways use mono- or oligosaccharides activated by a diphosphate linkage to lipids, such as dolichol. These activated donors are then substrates for enzymes known as glycosyltransferases, which transfer the sugar unit from the activated donor to an accepting nucleophile (the acceptor substrate).

Disaccharide phosphorylases
Different biocatalytic approaches have been developed toward the synthesis of glycosides in the past decades, which using “glycosyltransferases” and “glycoside hydrolases” are among the most common catalysis. The former often needs expensive materials and the later often shows low yields, De Winter et al. investigated use of cellobiose phosphorylase (CP) toward synthesis of alpha-glycosides in ionic liquids. The best condition for use of CP was found to be in the presence of IL AMMOENG 101 and ethyl acetate.
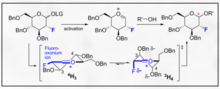
Directed glycosylations
Multiple chemical approaches exist to encourage selectivity of α- and β-glycosidic bonds. The highly substrate specific nature of the selectivity and the overall activity of the pyranoside can provide major synthetic difficulties. The overall specificity of the glycosylation can be improved by utilizing approaches which take into account the relative transition states that the anomeric carbon can undergo during a typical glycosylation. Most notably, recognition and incorporation of Felkin-Ahn-Eisenstein models into rationale chemical design can generally provide reliable results provided the transformation can undergo this type of conformational control in the transition state.
Fluorine directed glycosylations represent an encouraging handle for both B selectivity and introduction of a non-natural biomimetic C2 functionality on the carbohydrate. One innovative example provided by Bucher et al. provides a way to utilize a fluoro oxonium ion and the trichloroacetimidate to encourage B stereoselectivity through the gauche effect. This reasonable stereoselectivity is clear through visualization of the Felkin-Ahn models of the possible chair forms.
This method represents an encouraging way to selectivity incorporate B-ethyl, isopropyl and other glycosides with typical trichloroacetimidate chemistry.
O-linked glycopeptides; pharmaceutical uses of O-glycosylated peptides
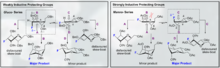
O-linked glycopeptides recently have been shown to exhibit excellent CNS permeability and efficacy in multiple animal models with disease states. In addition one of the most intriguing aspects thereof is the capability of O-glycosylation to extend half life, decrease clearance, and improve PK/PD thereof the active peptide beyond increasing CNS penetration. The innate utilization of sugars as solubilizing moieties in Phase II and III metabolism (glucuronic acids) has remarkably allowed an evolutionary advantage in that mammalian enzymes are not directly evolved to degrade O glycosylated products on larger moieties.
The peculiar nature of O-linked glycopeptides is that there are numerous examples which are CNS penetrant. The fundamental basis of this effect is thought to involve "membrane hopping" or "hop diffusion". The non-brownian motion driven "hop diffusion" process is thought to occur due to discontinuity of the plasma membrane. "Hop diffusion" notably combines free diffusion and intercomparmental transitions. Recent examples notably include high permeability of met-enkephalin analogs amongst other peptides. The full mOR agonist pentapeptide DAMGO is also CNS penetrant upon introduction of glycosylation.
N-Glycosidic bonds in DNA
DNA molecules contain 5-membered carbon rings called riboses that are directly attached to two phosphate groups and a nucleobase that contains amino groups. The nitrogen atoms from the amino group in the nucleotides are covalently linked to the anomeric carbon of the ribose sugar structure through an N-glycosidic bond. Occasionally, the nucleobases attached to the ribose undergo deamination, alkylation, or oxidation which results in cytotoxic lesions along the DNA backbone. These modifications severely threaten the cohesiveness of the DNA molecule, leading to the development of diseases such as cancer. DNA glycosylases are enzymes that catalyze the hydrolysis the N-glycosidic bond to free the damaged or modified nucleobase from the DNA, by cleaving the carbon-nitrogen glycosidic bond at the 2' carbon, subsequently initiating the base excision repair (BER) pathway.
Monofunctional glycosylases catalyze the hydrolysis of the N-glycosidic bond via either a stepwise, SN1 like mechanism, or a concerted, SN2 like mechanism. The stepwise function, the nucleobase acts as a leaving group before the anomeric carbon gets attacked by the water molecule, producing a short-lived unstable oxacarbenium ion intermediate. This intermediate rapidly reacts with the nearby water molecule to substitute the N-glycosidic bond of the ribose and the nucleobase with an O-glycosidic bond with a hydroxy group. The concerted mechanism, the water acts as a nucleophile and attacks at the anomeric carbon before the nucelobase gets to act like a leaving group. The intermediate produced is a similar oxacarbenium ion where both the hydroxy groups and the nucleobase are still attached to the anomeric carbon. Both mechanisms theoretically yield the same product. Most ribonucleotides are hydrolyzed via the concerted SN2 like mechanism, while most deoxyribonucleotides proceed through the stepwise like mechanism.
These reactions are practically irreversible. Due to the fact that the cleavage of the N-glycosidic bond from the DNA backbone can lead to detrimental mutagenic and cytotoxic responses in an organism, have the ability to also catalyze the synthesis of N-glycosidic bonds by way of an abasic DNA site and a specific nucleobase.
- Marco Brito-Arias, "Synthesis and Characterization of Glycosides", second edition, Editorial Springer 2016.
External links
- Definition of glycosides, from the IUPAC Compendium of Chemical Terminology, the "Gold Book"
- Varki A et al. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; 1999. Searchable online
|
Intramolecular (strong) |
|
|
||||||
|---|---|---|---|---|---|---|---|---|
|
Intermolecular (weak) |
|
|||||||
| Bond cleavage | ||||||||
| Electron counting rules | ||||||||
|
Types of carbohydrates
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| General | |||||||||||||||
| Geometry | |||||||||||||||
| Monosaccharides |
|
||||||||||||||
| Multiple |
|
||||||||||||||