
H3K9me2 is an epigenetic modification to the DNA packaging protein Histone H3. It is a mark that indicates the di-methylation at the 9th lysine residue of the histone H3 protein. H3K9me2 is strongly associated with transcriptional repression. H3K9me2 levels are higher at silent compared to active genes in a 10kb region surrounding the transcriptional start site. H3K9me2 represses gene expression both passively, by prohibiting acetylation as therefore binding of RNA polymerase or its regulatory factors, and actively, by recruiting transcriptional repressors. H3K9me2 has also been found in megabase blocks, termed Large Organised Chromatin K9 domains (LOCKS), which are primarily located within gene-sparse regions but also encompass genic and intergenic intervals. Its synthesis is catalyzed by G9a, G9a-like protein, and PRDM2. H3K9me2 can be removed by a wide range of histone lysine demethylases (KDMs) including KDM1, KDM3, KDM4 and KDM7 family members. H3K9me2 is important for various biological processes including cell lineage commitment, the reprogramming of somatic cells to induced pluripotent stem cells, regulation of the inflammatory response, and addiction to drug use.
Nomenclature
H3K9me2 indicates dimethylation of lysine 9 on histone H3 protein subunit:
| Abbr. | Meaning |
| H3 | H3 family of histones |
| K | standard abbreviation for lysine |
| 9 | position of amino acid residue
(counting from N-terminus) |
| me | methyl group |
| 2 | number of methyl groups added |
Lysine Methylation
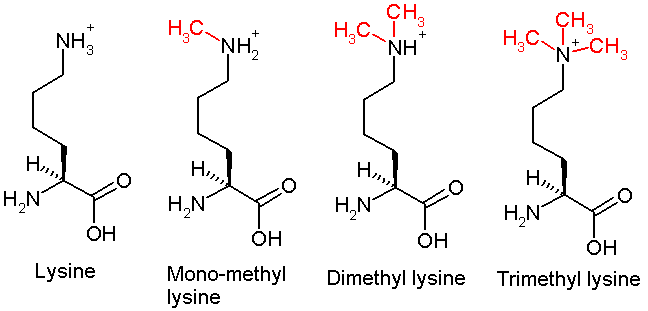
This diagram shows the progressive methylation of a lysine residue. The di-methylation denotes the methylation present in H3K9me2.
Understanding histone modifications
The genomic DNA of eukaryotic cells is wrapped around special protein molecules known as histones. The complexes formed by the looping of the DNA are known as chromatin. The basic structural unit of chromatin is the nucleosome: this consists of the core octamer of histones (H2A, H2B, H3 and H4) as well as a linker histone and about 180 base pairs of DNA. These core histones are rich in lysine and arginine residues. The carboxyl (C) terminal end of these histones contribute to histone-histone interactions, as well as histone-DNA interactions. The amino (N) terminal charged tails are the site of the post-translational modifications, such as the one seen in H3K9me2.
Epigenetic implications
The post-translational modification of histone tails by either histone modifying complexes or chromatin remodelling complexes are interpreted by the cell and lead to complex, combinatorial transcriptional output. It is thought that a histone code dictates the expression of genes by a complex interaction between the histones in a particular region. The current understanding and interpretation of histones comes from two large scale projects: ENCODE and the Epigenomic roadmap. The purpose of the epigenomic study was to investigate epigenetic changes across the entire genome. This led to chromatin states which define genomic regions by grouping the interactions of different proteins and/or histone modifications together. Chromatin states were investigated in Drosophila cells by looking at the binding location of proteins in the genome. Use of chromatin immunoprecipitation (ChIP)-sequencing revealed regions in the genome characterised by different banding. Different developmental stages were profiled in Drosophila as well, an emphasis was placed on histone modification relevance. A look in to the data obtained led to the definition of chromatin states based on histone modifications. Certain modifications were mapped and enrichment was seen to localize in certain genomic regions. Five core histone modifications were found with each respective one being linked to various cell functions.
- H3K4me3-promoters
- H3K4me1- primed enhancers
- H3K36me3-gene bodies
- H3K27me3-polycomb repression
- H3K9me3-heterochromatin
- H3K9me2-facultative heterochromatin
The human genome was annotated with chromatin states. These annotated states can be used as new ways to annotate a genome independently of the underlying genome sequence. This independence from the DNA sequence enforces the epigenetic nature of histone modifications. Chromatin states are also useful in identifying regulatory elements that have no defined sequence, such as enhancers. This additional level of annotation allows for a deeper understanding of cell specific gene regulation.
Clinical significance
Addiction
Chronic addictive drug exposure results in ΔFosB-mediated repression of G9a and reduced H3K9 dimethylation in the nucleus accumbens, which in turn causes dendritic arborization, altered synaptic protein expression, and increased drug seeking behavior. In contrast, accumbal G9a hyperexpression results in markedly increased H3K9 dimethylation and blocks the induction of this neural and behavioral plasticity by chronic drug use, which occurs via H3K9me2-mediated repression of transcription factors for ΔFosB and H3K9me2-mediated repression of various ΔFosB transcriptional targets (e.g., CDK5). Due to the involvement of H3K9me2 in these feedback loops and the central pathophysiological role of ΔFosB overexpression as the mechanistic trigger for addiction, the reduction of accumbal H3K9me2 following repeated drug exposure directly mediates the development of drug addictions.
Friedreich's ataxia
R-loop's are found with H3K9me2 mark at FXN in Friedreich's ataxia cells.
Cardiovascular disease
H3K9me2 is present at a subset of cardiovascular disease-associated gene promoters in vascular smooth muscle cells to block binding of NFκB and AP-1 (activator protein-1) transcription factors. Reduced levels of H3K9me2 have been observed in vascular smooth muscle cells from human atherosclerotic lesions compared to healthy aortic tissue in patients. Vascular smooth muscle cells from diabetic patients display reduced levels of H3K9me2 compared to non-diabetic controls; it has therefore been suggested that dysregulation of H3K9me2 might underlie the vascular complications associated with diabetes. Loss of H3K9me2 in vascular smooth muscle cells exacerbates upregulation of a subset of cardiovascular disease-associated genes in vascular disease models.
Methods
Histone modifications, including H3K9me2, can be detected using a variety of methods:
- Chromatin Immunoprecipitation Sequencing (ChIP-sequencing) measures the amount of DNA enrichment once bound to a targeted protein and immunoprecipitated. It results in good optimization and is used in vivo to reveal DNA-protein binding occurring in cells. ChIP-Seq can be used to identify and quantify various DNA fragments for different histone modifications along a genomic region.
- CUT&RUN(Cleavage Under Targets and Release Using Nuclease). In CUT&RUN, targeted DNA-protein complexes are isolated directly from the cell nucleus rather than following a precipitation step. To perform CUT&RUN, a specific antibody to the DNA-binding protein of interest and ProtA-MNase is added to permeabilised cells. MNase is tethered to the protein of interest through the ProtA-antibody interaction and MNase cleaves the surrounding, unprotected DNA to release protein-DNA complexes, which can then be isolated and sequenced. CUT&RUN is reported to give a much higher signal to noise ratio compared to traditional ChIP. CUT&RUN therefore requires one tenth of the sequencing depth of ChIP and permits genomic mapping of histone modifications and transcription factors using extremely low cell numbers.
- Modification-specific intracellular antibody probes. Sensitive fluorescent genetically encoded histone modification-specific intracellular antibody (mintbody) probes can be used to monitor changes in histone modifications in living cells.