
In biology, histones are highly basic proteins abundant in lysine and arginine residues that are found in eukaryotic cell nuclei. They act as spools around which DNA winds to create structural units called nucleosomes. Nucleosomes in turn are wrapped into 30-nanometer fibers that form tightly packed chromatin. Histones prevent DNA from becoming tangled and protect it from DNA damage. In addition, histones play important roles in gene regulation and DNA replication. Without histones, unwound DNA in chromosomes would be very long. For example, each human cell has about 1.8 meters of DNA if completely stretched out; however, when wound about histones, this length is reduced to about 90 micrometers (0.09 mm) of 30 nm diameter chromatin fibers.
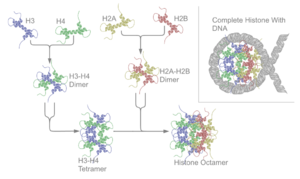
There are five families of histones which are designated H1/H5 (linker histones), H2, H3, and H4 (core histones). The nucleosome core is formed of two H2A-H2B dimers and a H3-H4 tetramer. The tight wrapping of DNA around histones is to a large degree a result of electrostatic attraction between the positively charged histones and negatively charged phosphate backbone of DNA.
Histones may be chemically modified through the action of enzymes to regulate gene transcription. The most common modification are the methylation of arginine or lysine residues or the acetylation of lysine. Methylation can affect how other protein such as transcription factors interact with the nucleosomes. Lysine acetylation eliminates a positive charge on lysine thereby weakening the electrostatic attraction between histone and DNA resulting in partial unwinding of the DNA making it more accessible for gene expression.
Classes and variants

Five major families of histones exist: H1/H5, H2A, H2B, H3, and H4. Histones H2A, H2B, H3 and H4 are known as the core or nucleosomal histones, while histones H1/H5 are known as the linker histones.
The core histones all exist as dimers, which are similar in that they all possess the histone fold domain: three alpha helices linked by two loops. It is this helical structure that allows for interaction between distinct dimers, particularly in a head-tail fashion (also called the handshake motif). The resulting four distinct dimers then come together to form one octameric nucleosome core, approximately 63 Angstroms in diameter (a solenoid (DNA)-like particle). Around 146 base pairs (bp) of DNA wrap around this core particle 1.65 times in a left-handed super-helical turn to give a particle of around 100 Angstroms across. The linker histone H1 binds the nucleosome at the entry and exit sites of the DNA, thus locking the DNA into place and allowing the formation of higher order structure. The most basic such formation is the 10 nm fiber or beads on a string conformation. This involves the wrapping of DNA around nucleosomes with approximately 50 base pairs of DNA separating each pair of nucleosomes (also referred to as linker DNA). Higher-order structures include the 30 nm fiber (forming an irregular zigzag) and 100 nm fiber, these being the structures found in normal cells. During mitosis and meiosis, the condensed chromosomes are assembled through interactions between nucleosomes and other regulatory proteins.
Histones are subdivided into canonical replication-dependent histones that are expressed during the S-phase of the cell cycle and replication-independent histone variants, expressed during the whole cell cycle. In animals, genes encoding canonical histones are typically clustered along the chromosome, lack introns and use a stem loop structure at the 3' end instead of a polyA tail. Genes encoding histone variants are usually not clustered, have introns and their mRNAs are regulated with polyA tails. Complex multicellular organisms typically have a higher number of histone variants providing a variety of different functions. Recent data are accumulating about the roles of diverse histone variants highlighting the functional links between variants and the delicate regulation of organism development. Histone variants from different organisms, their classification and variant specific features can be found in "HistoneDB 2.0 - Variants" database.
The following is a list of human histone proteins:
| Super family | Family | Subfamily | Members |
|---|---|---|---|
| Linker | H1 | H1F | H1F0, H1FNT, H1FOO, H1FX |
| H1H1 | HIST1H1A, HIST1H1B, HIST1H1C, HIST1H1D, HIST1H1E, HIST1H1T | ||
| Core | H2A | H2AF | H2AFB1, H2AFB2, H2AFB3, H2AFJ, H2AFV, H2AFX, H2AFY, H2AFY2, H2AFZ |
| H2A1 | HIST1H2AA, HIST1H2AB, HIST1H2AC, HIST1H2AD, HIST1H2AE, HIST1H2AG, HIST1H2AI, HIST1H2AJ, HIST1H2AK, HIST1H2AL, HIST1H2AM | ||
| H2A2 | HIST2H2AA3, HIST2H2AC | ||
| H2B | H2BF | H2BFM, H2BC12L, H2BFWT | |
| H2B1 | HIST1H2BA, HIST1H2BB, HIST1H2BC, HIST1H2BD, HIST1H2BE, HIST1H2BF, HIST1H2BG, HIST1H2BH, HIST1H2BI, HIST1H2BJ, HIST1H2BK, HIST1H2BL, HIST1H2BM, HIST1H2BN, HIST1H2BO | ||
| H2B2 | HIST2H2BE | ||
| H3 | H3A1 | HIST1H3A, HIST1H3B, HIST1H3C, HIST1H3D, HIST1H3E, HIST1H3F, HIST1H3G, HIST1H3H, HIST1H3I, HIST1H3J | |
| H3A2 | HIST2H3C | ||
| H3A3 | HIST3H3 | ||
| H4 | H41 | HIST1H4A, HIST1H4B, HIST1H4C, HIST1H4D, HIST1H4E, HIST1H4F, HIST1H4G, HIST1H4H, HIST1H4I, HIST1H4J, HIST1H4K, HIST1H4L | |
| H44 | HIST4H4 |
Structure

The nucleosome core is formed of two H2A-H2B dimers and a H3-H4 tetramer, forming two nearly symmetrical halves by tertiary structure (C2 symmetry; one macromolecule is the mirror image of the other). The H2A-H2B dimers and H3-H4 tetramer also show pseudodyad symmetry. The 4 'core' histones (H2A, H2B, H3 and H4) are relatively similar in structure and are highly conserved through evolution, all featuring a 'helix turn helix turn helix' motif (DNA-binding protein motif that recognize specific DNA sequence). They also share the feature of long 'tails' on one end of the amino acid structure - this being the location of post-translational modification (see below).
Archaeal histone only contains a H3-H4 like dimeric structure made out of a single type of unit. Such dimeric structures can stack into a tall superhelix ("hypernucleosome") onto which DNA coils in a manner similar to nucleosome spools. Only some archaeal histones have tails.
The distance between the spools around which eukaryotic cells wind their DNA has been determined to range from 59 to 70 Å.
In all, histones make five types of interactions with DNA:
- Salt bridges and hydrogen bonds between side chains of basic amino acids (especially lysine and arginine) and phosphate oxygens on DNA
- Helix-dipoles form alpha-helixes in H2B, H3, and H4 cause a net positive charge to accumulate at the point of interaction with negatively charged phosphate groups on DNA
- Hydrogen bonds between the DNA backbone and the amide group on the main chain of histone proteins
- Nonpolar interactions between the histone and deoxyribose sugars on DNA
- Non-specific minor groove insertions of the H3 and H2B N-terminal tails into two minor grooves each on the DNA molecule
The highly basic nature of histones, aside from facilitating DNA-histone interactions, contributes to their water solubility.
Histones are subject to post translational modification by enzymes primarily on their N-terminal tails, but also in their globular domains. Such modifications include methylation, citrullination, acetylation, phosphorylation, SUMOylation, ubiquitination, and ADP-ribosylation. This affects their function of gene regulation.
In general, genes that are active have less bound histone, while inactive genes are highly associated with histones during interphase. It also appears that the structure of histones has been evolutionarily conserved, as any deleterious mutations would be severely maladaptive. All histones have a highly positively charged N-terminus with many lysine and arginine residues.
Evolution and species distribution
Core histones are found in the nuclei of eukaryotic cells and in most Archaeal phyla, but not in bacteria. The unicellular algae known as dinoflagellates were previously thought to be the only eukaryotes that completely lack histones, but later studies showed that their DNA still encodes histone genes. Unlike the core histones, homologs of the lysine-rich linker histone (H1) proteins are found in bacteria, otherwise known as nucleoprotein HC1/HC2.
It has been proposed that core histone proteins are evolutionarily related to the helical part of the extended AAA+ ATPase domain, the C-domain, and to the N-terminal substrate recognition domain of Clp/Hsp100 proteins. Despite the differences in their topology, these three folds share a homologous helix-strand-helix (HSH) motif. It’s also proposed that they may have evolved from ribosomal proteins (RPS6/RPS15), both being short and basic proteins.
Archaeal histones may well resemble the evolutionary precursors to eukaryotic histones. Histone proteins are among the most highly conserved proteins in eukaryotes, emphasizing their important role in the biology of the nucleus. In contrast mature sperm cells largely use protamines to package their genomic DNA, most likely because this allows them to achieve an even higher packaging ratio.
There are some variant forms in some of the major classes. They share amino acid sequence homology and core structural similarity to a specific class of major histones but also have their own feature that is distinct from the major histones. These minor histones usually carry out specific functions of the chromatin metabolism. For example, histone H3-like CENPA is associated with only the centromere region of the chromosome. Histone H2A variant H2A.Z is associated with the promoters of actively transcribed genes and also involved in the prevention of the spread of silent heterochromatin. Furthermore, H2A.Z has roles in chromatin for genome stability. Another H2A variant H2A.X is phosphorylated at S139 in regions around double-strand breaks and marks the region undergoing DNA repair. Histone H3.3 is associated with the body of actively transcribed genes.
Function

Compacting DNA strands
Histones act as spools around which DNA winds. This enables the compaction necessary to fit the large genomes of eukaryotes inside cell nuclei: the compacted molecule is 40,000 times shorter than an unpacked molecule.
Chromatin regulation
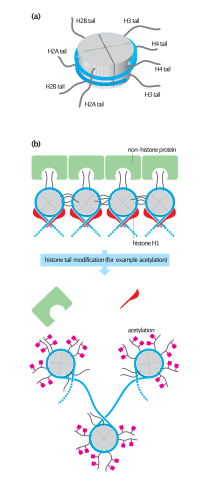
Histones undergo posttranslational modifications that alter their interaction with DNA and nuclear proteins. The H3 and H4 histones have long tails protruding from the nucleosome, which can be covalently modified at several places. Modifications of the tail include methylation, acetylation, phosphorylation, ubiquitination, SUMOylation, citrullination, and ADP-ribosylation. The core of the histones H2A and H2B can also be modified. Combinations of modifications, known as histone marks, are thought to constitute a code, the so-called "histone code". Histone modifications act in diverse biological processes such as gene regulation, DNA repair, chromosome condensation (mitosis) and spermatogenesis (meiosis).
The common nomenclature of histone modifications is:
- The name of the histone (e.g., H3)
- The single-letter amino acid abbreviation (e.g., K for Lysine) and the amino acid position in the protein
- The type of modification (Me: methyl, P: phosphate, Ac: acetyl, Ub: ubiquitin)
- The number of modifications (only Me is known to occur in more than one copy per residue. 1, 2 or 3 is mono-, di- or tri-methylation)
So H3K4me1 denotes the monomethylation of the 4th residue (a lysine) from the start (i.e., the N-terminal) of the H3 protein.
| Type of modification |
Histone | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| H3K4 | H3K9 | H3K14 | H3K27 | H3K79 | H3K36 | H4K20 | H2BK5 | H2BK20 | |
| mono-methylation | activation | activation | activation | activation | activation | activation | |||
| di-methylation | repression | repression | activation | ||||||
| tri-methylation | activation | repression | repression | activation, repression |
activation | repression | |||
| acetylation | activation | activation | activation | activation | activation | ||||
Modification

A huge catalogue of histone modifications have been described, but a functional understanding of most is still lacking. Collectively, it is thought that histone modifications may underlie a histone code, whereby combinations of histone modifications have specific meanings. However, most functional data concerns individual prominent histone modifications that are biochemically amenable to detailed study.
Chemistry
Lysine methylation
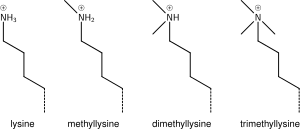
The addition of one, two, or many methyl groups to lysine has little effect on the chemistry of the histone; methylation leaves the charge of the lysine intact and adds a minimal number of atoms so steric interactions are mostly unaffected. However, proteins containing Tudor, chromo or PHD domains, amongst others, can recognise lysine methylation with exquisite sensitivity and differentiate mono, di and tri-methyl lysine, to the extent that, for some lysines (e.g.: H4K20) mono, di and tri-methylation appear to have different meanings. Because of this, lysine methylation tends to be a very informative mark and dominates the known histone modification functions.
Glutamine serotonylation
Recently it has been shown, that the addition of a serotonin group to the position 5 glutamine of H3, happens in serotonergic cells such as neurons. This is part of the differentiation of the serotonergic cells. This post-translational modification happens in conjunction with the H3K4me3 modification. The serotonylation potentiates the binding of the general transcription factor TFIID to the TATA box.
Arginine methylation
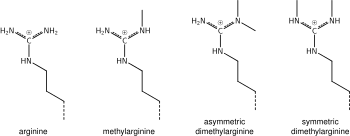
What was said above of the chemistry of lysine methylation also applies to arginine methylation, and some protein domains—e.g., Tudor domains—can be specific for methyl arginine instead of methyl lysine. Arginine is known to be mono- or di-methylated, and methylation can be symmetric or asymmetric, potentially with different meanings.
Arginine citrullination
Enzymes called peptidylarginine deiminases (PADs) hydrolyze the imine group of arginines and attach a keto group, so that there is one less positive charge on the amino acid residue. This process has been involved in the activation of gene expression by making the modified histones less tightly bound to DNA and thus making the chromatin more accessible. PADs can also produce the opposite effect by removing or inhibiting mono-methylation of arginine residues on histones and thus antagonizing the positive effect arginine methylation has on transcriptional activity.
Lysine acetylation
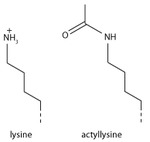
Addition of an acetyl group has a major chemical effect on lysine as it neutralises the positive charge. This reduces electrostatic attraction between the histone and the negatively charged DNA backbone, loosening the chromatin structure; highly acetylated histones form more accessible chromatin and tend to be associated with active transcription. Lysine acetylation appears to be less precise in meaning than methylation, in that histone acetyltransferases tend to act on more than one lysine; presumably this reflects the need to alter multiple lysines to have a significant effect on chromatin structure. The modification includes H3K27ac.
Serine/threonine/tyrosine phosphorylation
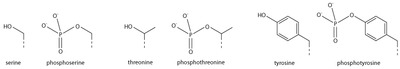
Addition of a negatively charged phosphate group can lead to major changes in protein structure, leading to the well-characterised role of phosphorylation in controlling protein function. It is not clear what structural implications histone phosphorylation has, but histone phosphorylation has clear functions as a post-translational modification, and binding domains such as BRCT have been characterised.
Effects on transcription
Most well-studied histone modifications are involved in control of transcription.
Actively transcribed genes
Two histone modifications are particularly associated with active transcription:
- Trimethylation of H3 lysine 4 (H3K4me3)
- This trimethylation occurs at the promoter of active genes and is performed by the COMPASS complex. Despite the conservation of this complex and histone modification from yeast to mammals, it is not entirely clear what role this modification plays. However, it is an excellent mark of active promoters and the level of this histone modification at a gene's promoter is broadly correlated with transcriptional activity of the gene. The formation of this mark is tied to transcription in a rather convoluted manner: early in transcription of a gene, RNA polymerase II undergoes a switch from initiating' to 'elongating', marked by a change in the phosphorylation states of the RNA polymerase II C terminal domain (CTD). The same enzyme that phosphorylates the CTD also phosphorylates the Rad6 complex, which in turn adds a ubiquitin mark to H2B K123 (K120 in mammals). H2BK123Ub occurs throughout transcribed regions, but this mark is required for COMPASS to trimethylate H3K4 at promoters.
- Trimethylation of H3 lysine 36 (H3K36me3)
- This trimethylation occurs in the body of active genes and is deposited by the methyltransferase Set2. This protein associates with elongating RNA polymerase II, and H3K36Me3 is indicative of actively transcribed genes. H3K36Me3 is recognised by the Rpd3 histone deacetylase complex, which removes acetyl modifications from surrounding histones, increasing chromatin compaction and repressing spurious transcription. Increased chromatin compaction prevents transcription factors from accessing DNA, and reduces the likelihood of new transcription events being initiated within the body of the gene. This process therefore helps ensure that transcription is not interrupted.
Repressed genes
Three histone modifications are particularly associated with repressed genes:
- Trimethylation of H3 lysine 27 (H3K27me3)
- This histone modification is deposited by the polycomb complex PRC2. It is a clear marker of gene repression, and is likely bound by other proteins to exert a repressive function. Another polycomb complex, PRC1, can bind H3K27me3 and adds the histone modification H2AK119Ub which aids chromatin compaction. Based on this data it appears that PRC1 is recruited through the action of PRC2, however, recent studies show that PRC1 is recruited to the same sites in the absence of PRC2.
- Di and tri-methylation of H3 lysine 9 (H3K9me2/3)
- H3K9me2/3 is a well-characterised marker for heterochromatin, and is therefore strongly associated with gene repression. The formation of heterochromatin has been best studied in the yeast Schizosaccharomyces pombe, where it is initiated by recruitment of the RNA-induced transcriptional silencing (RITS) complex to double stranded RNAs produced from centromeric repeats. RITS recruits the Clr4 histone methyltransferase which deposits H3K9me2/3. This process is called histone methylation. H3K9Me2/3 serves as a binding site for the recruitment of Swi6 (heterochromatin protein 1 or HP1, another classic heterochromatin marker) which in turn recruits further repressive activities including histone modifiers such as histone deacetylases and histone methyltransferases.
- Trimethylation of H4 lysine 20 (H4K20me3)
- This modification is tightly associated with heterochromatin, although its functional importance remains unclear. This mark is placed by the Suv4-20h methyltransferase, which is at least in part recruited by heterochromatin protein 1.
Bivalent promoters
Analysis of histone modifications in embryonic stem cells (and other stem cells) revealed many gene promoters carrying both H3K4Me3 and H3K27Me3, in other words these promoters display both activating and repressing marks simultaneously. This peculiar combination of modifications marks genes that are poised for transcription; they are not required in stem cells, but are rapidly required after differentiation into some lineages. Once the cell starts to differentiate, these bivalent promoters are resolved to either active or repressive states depending on the chosen lineage.
Other functions
DNA damage repair
Marking sites of DNA damage is an important function for histone modifications. Without a repair marker, DNA would get destroyed by damage accumulated from sources such as the ultraviolet radiation of the sun.
- Phosphorylation of H2AX at serine 139 (γH2AX)
- Phosphorylated H2AX (also known as gamma H2AX) is a marker for DNA double strand breaks, and forms part of the response to DNA damage. H2AX is phosphorylated early after detection of DNA double strand break, and forms a domain extending many kilobases either side of the damage. Gamma H2AX acts as a binding site for the protein MDC1, which in turn recruits key DNA repair proteins (this complex topic is well reviewed in) and as such, gamma H2AX forms a vital part of the machinery that ensures genome stability.
- Acetylation of H3 lysine 56 (H3K56Ac)
- H3K56Acx is required for genome stability. H3K56 is acetylated by the p300/Rtt109 complex, but is rapidly deacetylated around sites of DNA damage. H3K56 acetylation is also required to stabilise stalled replication forks, preventing dangerous replication fork collapses. Although in general mammals make far greater use of histone modifications than microorganisms, a major role of H3K56Ac in DNA replication exists only in fungi, and this has become a target for antibiotic development.
- Trimethylation of H3 lysine 36 (H3K36me3)
- H3K36me3 has the ability to recruit the MSH2-MSH6 (hMutSα) complex of the DNA mismatch repair pathway. Consistently, regions of the human genome with high levels of H3K36me3 accumulate less somatic mutations due to mismatch repair activity.
Chromosome condensation
- Phosphorylation of H3 at serine 10 (phospho-H3S10)
- The mitotic kinase aurora B phosphorylates histone H3 at serine 10, triggering a cascade of changes that mediate mitotic chromosome condensation. Condensed chromosomes therefore stain very strongly for this mark, but H3S10 phosphorylation is also present at certain chromosome sites outside mitosis, for example in pericentric heterochromatin of cells during G2. H3S10 phosphorylation has also been linked to DNA damage caused by R-loop formation at highly transcribed sites.
- Phosphorylation H2B at serine 10/14 (phospho-H2BS10/14)
- Phosphorylation of H2B at serine 10 (yeast) or serine 14 (mammals) is also linked to chromatin condensation, but for the very different purpose of mediating chromosome condensation during apoptosis. This mark is not simply a late acting bystander in apoptosis as yeast carrying mutations of this residue are resistant to hydrogen peroxide-induced apoptotic cell death.
Addiction
Epigenetic modifications of histone tails in specific regions of the brain are of central importance in addictions. Once particular epigenetic alterations occur, they appear to be long lasting "molecular scars" that may account for the persistence of addictions.
Cigarette smokers (about 15% of the US population) are usually addicted to nicotine. After 7 days of nicotine treatment of mice, acetylation of both histone H3 and histone H4 was increased at the FosB promoter in the nucleus accumbens of the brain, causing 61% increase in FosB expression. This would also increase expression of the splice variant Delta FosB. In the nucleus accumbens of the brain, Delta FosB functions as a "sustained molecular switch" and "master control protein" in the development of an addiction.
About 7% of the US population is addicted to alcohol. In rats exposed to alcohol for up to 5 days, there was an increase in histone 3 lysine 9 acetylation in the pronociceptin promoter in the brain amygdala complex. This acetylation is an activating mark for pronociceptin. The nociceptin/nociceptin opioid receptor system is involved in the reinforcing or conditioning effects of alcohol.
Methamphetamine addiction occurs in about 0.2% of the US population. Chronic methamphetamine use causes methylation of the lysine in position 4 of histone 3 located at the promoters of the c-fos and the C-C chemokine receptor 2 (ccr2) genes, activating those genes in the nucleus accumbens (NAc). c-fos is well known to be important in addiction. The ccr2 gene is also important in addiction, since mutational inactivation of this gene impairs addiction.
Synthesis
The first step of chromatin structure duplication is the synthesis of histone proteins: H1, H2A, H2B, H3, H4. These proteins are synthesized during S phase of the cell cycle. There are different mechanisms which contribute to the increase of histone synthesis.
Yeast
Yeast carry one or two copies of each histone gene, which are not clustered but rather scattered throughout chromosomes. Histone gene transcription is controlled by multiple gene regulatory proteins such as transcription factors which bind to histone promoter regions. In budding yeast, the candidate gene for activation of histone gene expression is SBF. SBF is a transcription factor that is activated in late G1 phase, when it dissociates from its repressor Whi5. This occurs when Whi5 is phosphorylated by Cdc8 which is a G1/S Cdk. Suppression of histone gene expression outside of S phases is dependent on Hir proteins which form inactive chromatin structure at the locus of histone genes, causing transcriptional activators to be blocked.
Metazoan
In metazoans the increase in the rate of histone synthesis is due to the increase in processing of pre-mRNA to its mature form as well as decrease in mRNA degradation; this results in an increase of active mRNA for translation of histone proteins. The mechanism for mRNA activation has been found to be the removal of a segment of the 3' end of the mRNA strand, and is dependent on association with stem-loop binding protein (SLBP). SLBP also stabilizes histone mRNAs during S phase by blocking degradation by the 3'hExo nuclease. SLBP levels are controlled by cell-cycle proteins, causing SLBP to accumulate as cells enter S phase and degrade as cells leave S phase. SLBP are marked for degradation by phosphorylation at two threonine residues by cyclin dependent kinases, possibly cyclin A/ cdk2, at the end of S phase. Metazoans also have multiple copies of histone genes clustered on chromosomes which are localized in structures called Cajal bodies as determined by genome-wide chromosome conformation capture analysis (4C-Seq).
Link between cell-cycle control and synthesis
Nuclear protein Ataxia-Telangiectasia (NPAT), also known as nuclear protein coactivator of histone transcription, is a transcription factor which activates histone gene transcription on chromosomes 1 and 6 of human cells. NPAT is also a substrate of cyclin E-Cdk2, which is required for the transition between G1 phase and S phase. NPAT activates histone gene expression only after it has been phosphorylated by the G1/S-Cdk cyclin E-Cdk2 in early S phase. This shows an important regulatory link between cell-cycle control and histone synthesis.
History
Histones were discovered in 1884 by Albrecht Kossel. The word "histone" dates from the late 19th century and is derived from the German word "Histon", a word itself of uncertain origin, perhaps from Ancient Greek ἵστημι (hístēmi, “make stand”) or ἱστός (histós, “loom”).
In the early 1960s, before the types of histones were known and before histones were known to be highly conserved across taxonomically diverse organisms, James F. Bonner and his collaborators began a study of these proteins that were known to be tightly associated with the DNA in the nucleus of higher organisms. Bonner and his postdoctoral fellow Ru Chih C. Huang showed that isolated chromatin would not support RNA transcription in the test tube, but if the histones were extracted from the chromatin, RNA could be transcribed from the remaining DNA. Their paper became a citation classic. Paul T'so and James Bonner had called together a World Congress on Histone Chemistry and Biology in 1964, in which it became clear that there was no consensus on the number of kinds of histone and that no one knew how they would compare when isolated from different organisms. Bonner and his collaborators then developed methods to separate each type of histone, purified individual histones, compared amino acid compositions in the same histone from different organisms, and compared amino acid sequences of the same histone from different organisms in collaboration with Emil Smith from UCLA. For example, they found Histone IV sequence to be highly conserved between peas and calf thymus. However, their work on the biochemical characteristics of individual histones did not reveal how the histones interacted with each other or with DNA to which they were tightly bound.
Also in the 1960s, Vincent Allfrey and Alfred Mirsky had suggested, based on their analyses of histones, that acetylation and methylation of histones could provide a transcriptional control mechanism, but did not have available the kind of detailed analysis that later investigators were able to conduct to show how such regulation could be gene-specific. Until the early 1990s, histones were dismissed by most as inert packing material for eukaryotic nuclear DNA, a view based in part on the models of Mark Ptashne and others, who believed that transcription was activated by protein-DNA and protein-protein interactions on largely naked DNA templates, as is the case in bacteria.
During the 1980s, Yahli Lorch and Roger Kornberg showed that a nucleosome on a core promoter prevents the initiation of transcription in vitro, and Michael Grunstein demonstrated that histones repress transcription in vivo, leading to the idea of the nucleosome as a general gene repressor. Relief from repression is believed to involve both histone modification and the action of chromatin-remodeling complexes. Vincent Allfrey and Alfred Mirsky had earlier proposed a role of histone modification in transcriptional activation, regarded as a molecular manifestation of epigenetics. Michael Grunstein and David Allis found support for this proposal, in the importance of histone acetylation for transcription in yeast and the activity of the transcriptional activator Gcn5 as a histone acetyltransferase.
The discovery of the H5 histone appears to date back to the 1970s, and it is now considered an isoform of Histone H1.