
| Idiopathic multicentric Castleman disease | |
|---|---|
| Other names | Giant lymph node hyperplasia, lymphoid hamartoma, angiofollicular lymph node hyperplasia |
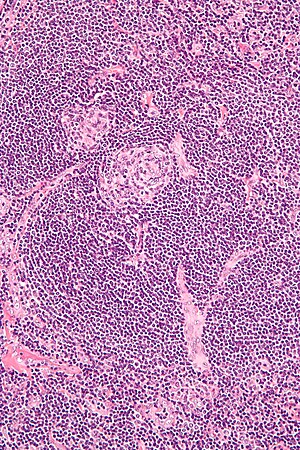 | |
| Micrograph of lymph node biopsy demonstrating hyaline vascular features consistent with Castleman disease | |
| Specialty | Hematology, immunology, rheumatology, pathology |
| Diagnostic method | Based on patient history, physical exam, laboratory testing, medical imaging, histopathology |
| Frequency | approximately 1500-1800 new cases per year in the United States |
Idiopathic multicentric Castleman disease (iMCD) is a subtype of Castleman disease (also known as giant lymph node hyperplasia, lymphoid hamartoma, or angiofollicular lymph node hyperplasia), a group of lymphoproliferative disorders characterized by lymph node enlargement, characteristic features on microscopic analysis of enlarged lymph node tissue, and a range of symptoms and clinical findings.
People with iMCD have enlarged lymph nodes in multiple regions and often have flu-like symptoms, abnormal findings on blood tests, and dysfunction of vital organs, such as the liver, kidneys, and bone marrow.
iMCD has features often found in autoimmune diseases and cancers, but the underlying disease mechanism is unknown. Treatment for iMCD may involve the use of a variety of medications, including immunosuppressants and chemotherapy.
Castleman disease was named after Dr. Benjamin Castleman, who first described the disease in 1956. The Castleman Disease Collaborative Network is the largest organization focused on the disease and is involved in research, awareness, and patient support.
Signs and symptoms
Patients with iMCD may experience enlarged lymph nodes in multiple lymph node regions; systemic symptoms (fever, night sweats, unintended weight loss, fatigue); enlargement of the liver and/or spleen; extravascular fluid accumulation in the extremities (edema), abdomen (ascites), or lining of the lungs (pleural effusion); lung symptoms such as cough and shortness of breath; and skin findings such as cherry hemangiomas.
Causes
The cause of iMCD is not known and no risk factors have been identified. Genetic variants have been observed in cases of Castleman disease; however, no genetic variant has been validated as disease causing.
Unlike HHV-8-associated MCD, iMCD is not caused by uncontrolled HHV-8 infection.
Mechanism
The disease mechanism of iMCD has not been fully described. It is known that interleukin-6 (IL-6), a molecule that stimulates immune cells, plays a role in some cases of iMCD. IL-6 levels measured in some patients with iMCD increase and decrease with corresponding changes in disease activity, mice treated with IL-6 develop features of iMCD, and blockade of the IL-6 pathway using the medications siltuximab and tocilizumab effectively treats some patients with iMCD. However, many patients with iMCD do not demonstrate elevated levels of IL-6 and IL-6 levels are not strongly correlated with response to treatment with anti-IL-6 medications. In cases where IL-6 does play a role, the underlying cause of elevated IL-6 levels and the cells responsible for producing IL-6 remain unknown.
Several theoretical mechanisms for iMCD have been proposed based on existing research and observed similarities between iMCD and other diseases that present with similar clinical findings and lymph node histology:
- Autoimmune – The immune system may produce antibodies that target healthy cells in the body instead of bacteria and viruses. Self-directed antibodies are commonly seen in autoimmune diseases such as systemic lupus erythematous and rheumatoid arthritis.
- Autoinflammatory – A mutation in a gene controlling inflammatory systems may contribute to harmful activation of inflammatory pathways in patients with iMCD.
- Neoplastic – Genetic mutations that develop in mature cells (somatic mutations) may cause an overgrowth of abnormal cells as in cancers such as lymphoma.
- Pathogen – Human herpesvirus 8 (HHV-8) is the known causative agent in HHV-8-associated MCD, which has very similar symptoms and findings to iMCD. While iMCD by definition is not caused by HHV-8, an unknown virus may cause the disease.
There have been no reported cases of UCD transforming into iMCD.
Diagnosis
iMCD is diagnosed according to evidence-based consensus diagnostic criteria, which require a thorough evaluation including patient history, physical exam, laboratory testing, radiologic imaging, and microscopic analysis (histology) of biopsied tissue from an enlarged lymph node. Diagnosis of iMCD requires clinical abnormalities, exclusion of other diseases, and a lymph node biopsy showing features consistent with Castleman disease. A lymph node biopsy alone is not sufficient to make the diagnosis.
Laboratory testing
Laboratory testing may demonstrate elevated C-reactive protein, decreased hemoglobin levels (anemia), low albumin levels, elevated creatinine, increased immunoglobulin levels, and abnormal (elevated or decreased) platelet counts. Patients may also have elevations of molecules involved in inflammation (cytokines), such as Interleukin 6 (IL-6) and vascular endothelial growth factor (VEGF).
Medical imaging
Radiologic imaging will demonstrate enlarged lymph nodes in multiple regions, which are typically 18F-fluorodoxyglucose (FDG) avid on positron-emission tomography (PET).
Associated diseases
iMCD is commonly seen in patients with POEMS syndrome, but it is unclear if iMCD occurs as an independent disease process or a manifestation of POEMS syndrome in these patients. Patients with iMCD have increased risk for solid tumors and cancers of the blood. Occasionally, patients with iMCD present with lymphocytic interstitial pneumonitis.
TAFRO Syndrome
iMCD patients with thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly syndrome (TAFRO syndrome) are considered to have a distinct clinical subtype of iMCD. Patients often have rapid progression of symptoms and frequently develop severe organ dysfunction. Compared to iMCD patients without TAFRO syndrome, iMCD patients with TAFRO syndrome are more likely to present with severe abdominal pain, low platelet levels, progressive renal dysfunction, and normal to mildly elevated immunoglobulin levels. While iMCD with TAFRO syndrome was first described in Japanese patients in 2010, cases of iMCD with TAFRO syndrome have since been reported in non-Japanese patients in many other countries.
Classification
Castleman disease describes a group of at least 3 distinct disorders—Unicentric Castleman disease (UCD), human herpesvirus 8 associated multicentric Castleman disease (HHV-8-associated MCD), and idiopathic multicentric Castleman disease (iMCD). Identifying the correct subtype of the disease is important, as the three disorders vary significantly in symptoms, clinical findings, disease mechanism, treatment approach, and prognosis.
- In Unicentric Castleman disease enlarged lymph nodes with characteristic microscopic findings are present in only a single lymph node region.
- In the multicentric subtypes of Castleman disease, enlarged lymph nodes with characteristic findings are present in multiple lymph node regions. The multicentric variants of Castleman disease are further classified by known causes of the disease.
- HHV-8-associated MCD is caused by uncontrolled infection with human herpesvirus 8 (HHV-8).
- In idiopathic multicentric Castleman disease (iMCD) the cause of the disease is unknown (idiopathic). Testing for HHV-8 must be negative to diagnose iMCD.
Idiopathic multicentric Castleman disease
iMCD may be further differentiated by the presence of associated diseases, such as polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes syndrome (POEMS syndrome), or by distinct clinical features, such as thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly syndrome (TAFRO syndrome).
Diagnostic criteria
Diagnosis of iMCD requires: the presence of both major criteria, multiple regions of enlarged lymph nodes as demonstrated by medical imaging; the presence of at least two minor criteria, at least one of which must be an abnormal laboratory test; and exclusion of diseases that can mimic iMCD.
Major criteria 1: multiple regions of enlarged lymph nodes
Radiologic imaging must demonstrate enlarged lymph nodes in multiple regions.
Major criteria 2: microscopic analysis of lymph node biopsy consistent with iMCD
The microscopic appearance (histology) of biopsied tissue from an enlarged lymph node must demonstrate a constellation of features consistent with Castleman disease. There are three patterns of characteristic histologic features associated with iMCD:
- Hypervascular - regressed germinal centers, follicular dendritic cell prominence, hypervascularity in interfollicular regions, and prominent mantle zones with an “onion-skin” appearance.
- Plasmacytic – increased number of follicles with large hyperplastic germinal centers and sheet-like plasmacytosis (increased number of plasma cells).
- Mixed – features of both hypervascular and plasmacytic.
iMCD most commonly demonstrates plasmacytic features; however, hypervascular features or a mixture of both hypervascular and plasmacytic features may also be seen in iMCD lymph nodes. The clinical utility of subtyping iMCD by histologic features is uncertain, as histologic subtypes do not consistently predict disease severity or treatment response.
Staining with latency-associated nuclear antigen (LANA-1), a marker of HHV-8 infection, must be negative to diagnose iMCD.
Minor criteria
Patients must experience at least two of the following 11 minor criteria with at least one being an abnormal laboratory test.
Laboratory tests:
- Elevated C-Reactive Protein or erythrocyte sedimentation rate
- Low hemoglobin levels (anemia)
- Abnormal (low or high) platelet counts
- Low albumin levels
- Elevated creatinine
- Increased levels of immunoglobulins (hypergammaglobulinemia)
Clinical features:
- Flu-like symptoms
- Enlargement of the liver and/or spleen
- Fluid accumulation (edema, ascites, pleural effusions)
- Skin findings such as cherry hemangiomas or violaceous papules
- Lymphocytic interstitial pneumonitis
Diseases to be excluded
Diagnosis requires exclusion of diseases that can present with similar clinical findings and similar appearance on microscopic analysis of tissue from an enlarged lymph node. Diseases that must be excluded in the diagnosis of iMCD include infectious diseases, such as HHV-8-associated MCD, Epstein-Barr virus mononucleosis, and reactive lymphadenopathy; autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis; and cancers, including lymphoma, multiple myeloma, and primary lymph node plasmacytoma.
Treatment
Due to the rarity of iMCD, data regarding treatment is limited and based on a combination of observational case series, case reports, and a single randomized clinical trial. Unlike UCD, for which surgery is the treatment of choice and curative for most patients, surgery is not effective in iMCD. Instead of surgical treatment, a variety of medications are used based on disease severity and a patient's response to prior treatments. Siltuximab, a monoclonal antibody targeting IL-6, is the only medication approved by the United States Food and Drug Administration (FDA) for the treatment of iMCD; however, successful use of other medications has been reported in the literature.
In 2018, the first evidence-based consensus treatment guidelines for iMCD were published by an international group of experts in the field. In addition to creating a treatment algorithm for iMCD, these treatment guidelines established common definitions for disease severity and response to treatment.
Evaluation of iMCD Severity
Patients with iMCD are classified as having severe or non-severe disease based on the 5 criteria listed below. Patient with 2 or more of the below criteria are classified as having severe disease while patients with 0-1 of the criteria are classified as having non-severe disease.
- Eastern Cooperative Oncology Group (ECOG) performance status ≥ 2
- Estimated glomerular filtration rate (eGFR) < 30 or Creatinine > 3.0 mg/dL
- Anasarca and/or ascites and/or pleural effusion and/or pericardial effusion
- Hemoglobin ≤ 8.0 g/dL
- Pulmonary involvement (e.g. interstitial pneumonitis with dyspnea)
Treatment response
Patients with iMCD are evaluated for treatment response based on changes in symptoms, sizes of involved lymph nodes, and laboratory testing. Each category is graded as a complete response, partial response, stable disease, or progressive disease. Overall treatment response is determined by the lowest category grade. For example, a patient with a complete laboratory response, a partial symptom response, and complete lymph node response would be given an overall treatment response of partial response. See below for descriptions of the criteria and grading of responses.
Laboratory Testing
Laboratory tests include all of the following: C-reactive protein, Hemoglobin, Albumin, and eGFR.
- Complete response - All lab values within normal ranges
- Partial response - >50% in all lab values
- Stable disease - All lab values between <50% improvement and <25% worsening
- Progressive disease - >25% worsening in any lab value
Symptoms
Four symptoms are assessed using the National Cancer Institute Common Terminology Criteria of Adverse Events (version 4): Fatigue, anorexia, fever, and body weight
- Complete response - Normalization to pre-disease baseline
- Partial response - Improvement in all 4 symptoms, but not to pre-disease baseline
- Stable disease - Improvement in at least 1 (but not all) symptoms
- Progressive disease - Worsening of at least 1 symptom on 2 or more assessments
Lymph Node
Treatment response for lymph nodes is evaluated using radiologic imaging and graded as complete response, partial response, stable disease, and progressive disease based on modified Cheson criteria.
Treatment Algorithm
The treatment algorithm for iMCD is based primarily on disease severity and response to treatment. Because of the high rate of relapse with withdrawal of treatment, most patients with iMCD are treated with medications indefinitely.
Non-severe disease
Siltuximab, an IL-6 blocker, is the recommended treatment for all patients with non-severe iMCD regardless of measured IL-6 levels. Tocilizumab, a drug that also targets the IL-6 pathway, is commonly used as an alternative to siltuximab when siltuximab is unavailable. Corticosteroids may be added to anti-IL-6 therapy depending on clinical presentation. Rituximab, a drug targeting B-cells, is primarily recommended as a second line therapy for patients who do not respond to siltuximab or tocilizumab, but may be used as a first line agent in appropriate patients.
For patients with non-severe disease who fail to respond to siltuximab, tocilizumab, and rituximab, treatment recommendations are not well defined. Cytotoxic chemotherapies have been reported to induce remission in patients with non-severe iMCD; however, the use of cytotoxic chemotherapies is not currently recommended for non-severe iMCD due to high likelihood of relapse and severe side effect profiles. As an alternative, immunomodulators such as thalidomide, cyclosporine A, sirolimus, bortezomib, and anakinra are recommended due to their similar response rates and more favorable long term side effect profiles.
Severe disease
Recommended initial treatment for all patients with severe iMCD is high dose steroids combined with an anti-IL-6 agent such as siltuximab or tocilizumab, regardless of measured IL-6 levels. For patients who immediately improve with this regimen, steroids may be slowly tapered, but the anti-IL-6 agent should be continued indefinitely due to the high relapse rate with withdrawal of treatment. Due to the high risk of complications associated with severe iMCD, if patients worsen or fail to improve with high dose steroids and anti-IL-6 therapies, cytotoxic chemotherapy regimens are recommended. Patients with life-threatening disease, particularly those with TAFRO Syndrome, may require advanced measures such as breathing support with a mechanical ventilator or treatment with dialysis for kidney failure.
Following improvement in disease status, maintenance therapy with an anti-IL-6 agent or an immunosuppressant medication is typically continued indefinitely, as withdrawal of such medications can lead to relapse.
Follow-up
Patients with iMCD require routine assessment of treatment response and disease progression. It is recommended that follow-up visits include evaluation of symptoms, physical examination, laboratory testing, and radiologic imaging.
Prognosis
iMCD can present as an acute life-threatening disease in some patients or a chronic disease in others. Some patients have longstanding stable disease while others experience flares of severe disease that may improve with treatment. Successful treatment controls symptoms and organ dysfunction associated with iMCD, improves symptoms and organ dysfunction during disease flares, and prevents future disease flares.
Observed survival in a recent study of iMCD patients was 92% at 2 years, 76% at 5 years, and 59% at 10 years.
Epidemiology
There are approximately 1500-1800 new cases of iMCD diagnosed per year in the United States. iMCD can occur at any age, but the median age at presentation is approximately 50 years old. There is a slightly increased incidence of iMCD in women.
There have been no published epidemiologic studies of Castleman disease outside of the United States; however, there has been no published data demonstrating increased or decreased incidence of Castleman disease in specific regions or ethnicities.
History
Castleman disease was first described by Dr. Benjamin Castleman in 1956. World Castleman Disease Day was established in 2018 and is held every year on July 23.
Culture
The Castleman Disease Collaborative Network was founded in 2012 and is the largest organization focused on Castleman disease. It is a global collaborative network involved in research, awareness, and patient support.