
 | |
| Clinical data | |
|---|---|
| Trade names | Mepact |
| License data | |
| Pregnancy category |
|
| Routes of administration |
intravenous liposomal infusion over one hour |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | N/A |
| Elimination half-life | minutes (in plasma) 18 hrs (terminal) |
| Identifiers | |
| |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
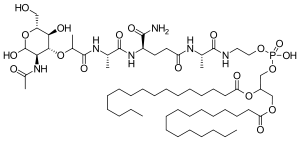
| Formula | C59H109N6O19P |
| Molar mass | 1237.518 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
|
| |
Mifamurtide (trade name Mepact, marketed by Takeda) is a drug against osteosarcoma, a kind of bone cancer mainly affecting children and young adults, which is lethal in over half of cases. The drug was approved in Europe in March 2009.
Medical uses
Mifamurtide is indicated for the treatment of high-grade, nonmetastasizing, resectable osteosarcoma following complete surgical removal in children, adolescents, and young adults, aged two to 30 years. Osteosarcoma is diagnosed in about 1,000 individuals in Europe and the USA per year, most under the age of 30. The drug is used in combination with postoperative, multiagent chemotherapy to kill remaining cancer cells and improve a patient's chance of overall survival.
In a phase-III clinical trial in about 800 newly diagnosed osteosarcoma patients, mifamurtide was combined with the chemotherapeutic agents doxorubicin and methotrexate, with or without cisplatin and ifosfamide. The mortality could be lowered by 30% versus chemotherapy plus placebo. Six years after the treatment, 78% of patients were still alive. This equals an absolute risk reduction of 8% .
Adverse effects
In a clinical study, mifamurtide was given to 332 subjects (half of whom were under age of 16) and most side effects were found to be mild to moderate in nature. Most patients experience fewer adverse events with subsequent administration. Common side effects include fever (about 90%), vomiting, fatigue and tachycardia (about 50%), infections, anaemia, anorexia, headache, diarrhoea and constipation (>10%).
Interactions
- Theoretical considerations suggest calcineurin inhibitors like ciclosporin and tacrolimus might interact with mifamurtide because of their effect on macrophages.
- High-dose NSAIDs block the mechanism of mifamurtide in vitro.
Consequently, the combination of mifamurtide with these types of drugs is contraindicated. However, mifamurtide can be coadministered with low doses of NSAIDs. No evidence suggests mifamurtide interacts with the studied chemotherapeutics, or with the cytochrome P450 system.
Pharmacology
Mechanism of action
Mifamurtide is a fully synthetic derivative of muramyl dipeptide (MDP), the smallest naturally occurring immune stimulatory component of cell walls from Mycobacterium species. It has similar immunostimulatory effects as natural MDP with the advantage of a longer half-life in plasma.
NOD2 is a pattern recognition receptor which is found in several kinds of white blood cells, mainly monocytes and macrophages. It recognises muramyl dipeptide, a component of the cell wall of bacteria. Mifamurtide simulates a bacterial infection by binding to NOD2, activating white cells. This results in an increased production of TNF-α, interleukin 1, interleukin 6, interleukin 8, interleukin 12, and other cytokines, as well as ICAM-1. The activated white cells attack cancer cells, but not, at least in vitro, other cells.
Pharmacokinetics
After application of the liposomal infusion, the drug is cleared from the plasma within minutes and is concentrated in lung, liver, spleen, nasopharynx, and thyroid. The terminal half-life is 18 hours. In patients receiving a second treatment after 11–12 weeks, no accumulation effects were observed.
Chemistry

Mifamurtide is muramyl tripeptide phosphatidylethanolamine (MTP-PE), a synthetic analogue of muramyl dipeptide. The side chains of the molecule give it a longer elimination half-life than the natural substance. The substance is applied encapsulated into liposomes (L-MTP-PE). Being a phospholipid, it accumulates in the lipid bilayer of the liposomes in the infusion.
Synthesis
One method of synthesis (shown first) is based on N,N'-dicyclohexylcarbodiimide (DCC) assisted esterification of N-acetylmuramyl-L-alanyl-D-isoglutaminyl-L-alanine with N-hydroxysuccinimide, followed by a condensation with 2-aminoethyl-2,3-dipalmitoylglycerylphosphoric acid in triethylamine (Et3N). A different approach (shown second) uses N-acetylmuramyl-L-alanyl-D-isoglutamine, hydroxysuccinimide and alanyl-2-aminoethyl-2,3-dipalmitoylglycerylphosphoric acid; that is, the alanine is introduced in the second step instead of the first.

|

|
History
The drug was invented by Ciba-Geigy (now Novartis) in the early 1980s and sold to Jenner Biotherapies in the 1990s. In 2003, IDM Pharma bought the rights and developed it further. IDM Pharma was acquired by Takeda along with mifamurtide in June 2009.
Mifamurtide had already been granted orphan drug status by the U.S. Food and Drug Administration (FDA) in 2001, and the European Medicines Agency (EMA) followed in 2004. It was approved in the 27 European Union member states plus Iceland, Liechtenstein, and Norway by a centralized marketing authorization in March 2009. The drug was denied approval by the FDA in 2007. Mifamurtide has been licensed by the EMA since March, 2009.