
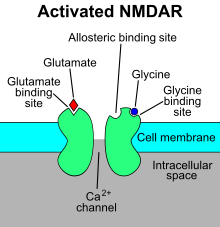
The N-methyl-D-aspartate receptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a “coincidence detector” and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.
The NMDA receptor is ionotropic, meaning it is a protein which allows the passage of ions through the cell membrane. The NMDA receptor is so named because the agonist molecule N-methyl-D-aspartate (NMDA) binds selectively to it, and not to other glutamate receptors. Activation of NMDA receptors results in the opening of the ion channel that is nonselective to cations, with a combined reversal potential near 0 mV. While the opening and closing of the ion channel is primarily gated by ligand binding, the current flow through the ion channel is voltage-dependent. Specifically located on the receptor, extracellular magnesium (Mg2+) and zinc (Zn2+) ions can bind and prevent other cations from flowing through the open ion channel. A voltage-dependent flow of sodium (Na+), calcium (Ca2+), and potassium (K+) ions into and out of the cell is made possible by the depolarization of the cell, which displaces and repels the Mg2+ and Zn2+ ions from the pore. Ca2+ flux through NMDA receptors in particular is thought to be critical in synaptic plasticity, a cellular mechanism for learning and memory, due to proteins which bind to and are activated by Ca2+ ions.
Activity of the NMDA receptor is blocked by many psychoactive drugs such as phencyclidine (PCP), alcohol (ethanol) and dextromethorphan (DXM). The anaesthetic and analgesic effects of the drugs ketamine and nitrous oxide are also partially due to their effects at blocking NMDA receptor activity. In contrast, overactivation of NMDAR by NMDA agonists increases the cytosolic concentrations of calcium and zinc, which significantly contributes to neural death, an effect known to be prevented by cannabinoids, mediated by activation of the CB1 receptor, which leads HINT1 protein to counteract the toxic effects of NMDAR-mediated NO production and zinc release. As well as preventing methamphetamine-induced neurotoxicity via inhibition of nitric oxide synthase (nNOS) expression and astrocyte activation, is seen to reduce methamphetamine induced brain damage through a CB1-dependent and independent mechanisms, respectively, and inhibition of methamphetamine induced astrogliosis is likely to occur through a CB2 receptor dependent mechanism for THC. Since 1989, memantine has been recognized to be an uncompetitive antagonist of the NMDA receptor, entering the channel of the receptor after it has been activated and thereby blocking the flow of ions.
Overactivation of the receptor, causing excessive influx of Ca2+ can lead to excitotoxicity which is implied to be involved in some neurodegenerative disorders. Blocking of NMDA receptors could therefore, in theory, be useful in treating such diseases. However, hypofunction of NMDA receptors (due to glutathione deficiency or other causes) may be involved in impairment of synaptic plasticity and could have other negative repercussions. The main problem with the utilization of NMDA receptor antagonists for neuroprotection is that the physiological actions of the NMDA receptor are essential for normal neuronal function. To be clinically useful NMDA antagonists need to block excessive activation without interfering with normal functions. Memantine has this property.
History
The discovery of NMDA receptors was followed by the synthesis and study of N-methyl-D-aspartic acid (NMDA) in the 1960s by Jeff Watkins and colleagues. In the early 1980s, NMDA receptors were shown to be involved in several central synaptic pathways. Receptor subunit selectivity was discovered in the early 1990s, which led to recognition of a new class of compounds that selectively inhibit the NR2B subunit. These findings led to vigorous campaign in the pharmaceutical industry. From this it was considered that NMDA receptors were associated with a variety of neurological disorders such as epilepsy, Parkinson's, Alzheimer's, Huntington's and other CNS disorders.
In 2002, it was discovered by Hilmar Bading and co-workers that the cellular consequences of NMDA receptor stimulation depend on the receptor's location on the neuronal cell surface. Synaptic NMDA receptors promote gene expression, plasticity-related events, and acquired neuroprotection. Extrasynaptic NMDA receptors promote death signaling; they cause transcriptional shut-off, mitochondrial dysfunction, and structural disintegration. This pathological triad of extrasynaptic NMDA receptor signaling represents a common conversion point in the etiology of several acute and chronic neurodegenerative conditions. The molecular basis for toxic extrasynaptic NMDA receptor signaling was uncovered by Hilmar Bading and co-workers in 2020.Extrasynaptic NMDA receptors form a death signaling complex with TRPM4. NMDAR/TRPM4 interaction interface inhibitors (also known as interface inhibitors) disrupt the NMDAR/TRPM4 complex and detoxify extrasynaptic NMDA receptors.
A fortuitous finding was made in 1968 when a woman was taking amantadine as flu medicine and experienced remarkable remission of her Parkinson's symptoms. This finding, reported by Scawab et al., was the beginning of medicinal chemistry of adamantane derivatives in the context of diseases affecting the CNS. Before this finding, memantine, another adamantane derivative, had been synthesized by Eli Lilly and Company in 1963. The purpose was to develop a hypoglycemic drug, but it showed no such efficacy. It was not until 1972 that a possible therapeutic importance of memantine for treating neurodegenerative disorders was discovered. From 1989 memantine has been recognized to be an uncompetitive antagonist of the NMDA receptor.
Structure

Functional NMDA receptors are heterotetramers composed of two GluN1 and typically two GluN2 subunits. There is one GluN1, four GluN2, and two GluN3 subunit encoding genes, and each gene may produce more than one splice variant.
- GluN1 – GRIN1
- GluN2
- GluN3
Gating

The NMDA receptor is a glutamate and ion channel protein receptor that is activated when glycine and glutamate bind to it. The receptor is a highly complex and dynamic heteromeric protein that interacts with a multitude of intracellular proteins via three distinct subunits, namely GluN1, GluN2, and GluN3. The GluN1 subunit, which is encoded by the GRIN1 gene, exhibits eight distinct isoforms owing to alternative splicing. On the other hand, the GluN2 subunit, of which there are four different types (A-D), as well as the GluN3 subunit, of which there are two types (A and B), are each encoded by six separate genes. This intricate molecular structure and genetic diversity enable the receptor to carry out a wide range of physiological functions within the nervous system. All the subunits share a common membrane topology that is dominated by a large extracellular N-terminus, a membrane region comprising three transmembrane segments, a re-entrant pore loop, an extracellular loop between the transmembrane segments that are structurally not well known, and an intracellular C-terminus, which are different in size depending on the subunit and provide multiple sites of interaction with many intracellular proteins. Figure 1 shows a basic structure of GluN1/GluN2 subunits that forms the binding site for memantine, Mg2+ and ketamine.
Mg2+ blocks the NMDA receptor channel in a voltage-dependent manner. The channels are also highly permeable to Ca2+. Activation of the receptor depends on glutamate binding, D-serine or glycine binding at its GluN1-linked binding site and AMPA receptor-mediated depolarization of the postsynaptic membrane, which relieves the voltage-dependent channel block by Mg2+. Activation and opening of the receptor channel thus allows the flow of K+, Na+ and Ca2+ ions, and the influx of Ca2+ triggers intracellular signaling pathways. Allosteric receptor binding sites for zinc, proteins and the polyamines spermidine and spermine are also modulators for the NMDA receptor channels.
The GluN2B subunit has been involved in modulating activity such as learning, memory, processing and feeding behaviors, as well as being implicated in number of human derangements. The basic structure and functions associated with the NMDA receptor can be attributed to the GluN2B subunit. For example, the glutamate binding site and the control of the Mg2+ block are formed by the GluN2B subunit. The high affinity sites for glycine antagonist are also exclusively displayed by the GluN1/GluN2B receptor.
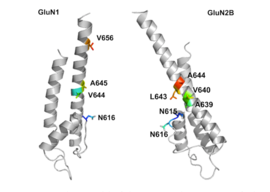
GluN1/GluN2B transmembrane segments are considered to be the part of the receptor that forms the binding pockets for uncompetitive NMDA receptor antagonists, but the transmembrane segments structures are not fully known as stated above. It is claimed that three binding sites within the receptor, A644 on the GluNB subunit and A645 and N616 on the GluN1 subunit, are important for binding of memantine and related compounds as seen in figure 2.
The NMDA receptor forms a heterotetramer between two GluN1 and two GluN2 subunits (the subunits were previously denoted as GluN1 and GluN2), two obligatory GluN1 subunits and two regionally localized GluN2 subunits. A related gene family of GluN3 A and B subunits have an inhibitory effect on receptor activity. Multiple receptor isoforms with distinct brain distributions and functional properties arise by selective splicing of the GluN1 transcripts and differential expression of the GluN2 subunits.
Each receptor subunit has modular design and each structural module, also represents a functional unit:
- The extracellular domain contains two globular structures: a modulatory domain and a ligand-binding domain. GluN1 subunits bind the co-agonist glycine and GluN2 subunits bind the neurotransmitter glutamate.
- The agonist-binding module links to a membrane domain, which consists of three transmembrane segments and a re-entrant loop reminiscent of the selectivity filter of potassium channels.
- The membrane domain contributes residues to the channel pore and is responsible for the receptor's high-unitary conductance, high-calcium permeability, and voltage-dependent magnesium block.
- Each subunit has an extensive cytoplasmic domain, which contain residues that can be directly modified by a series of protein kinases and protein phosphatases, as well as residues that interact with a large number of structural, adaptor, and scaffolding proteins.
The glycine-binding modules of the GluN1 and GluN3 subunits and the glutamate-binding module of the GluN2A subunit have been expressed as soluble proteins, and their three-dimensional structure has been solved at atomic resolution by x-ray crystallography. This has revealed a common fold with amino acid-binding bacterial proteins and with the glutamate-binding module of AMPA-receptors and kainate-receptors.
Mechanism of action
NMDA receptors are a crucial part of the development of the central nervous system. The processes of learning, memory, and neuroplasticity rely on the mechanism of NMDA receptors. NMDA receptors are glutamate-gated cation channels that allow for an increase of calcium permeability. Channel activation of NMDA receptors is a result of the binding of two co agonists, glycine and glutamate.
Overactivation of NMDA receptors, causing excessive influx of Ca2+ can lead to excitotoxicity. Excitotoxicity is implied to be involved in some neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease and Huntington's disease. Blocking of NMDA receptors could therefore, in theory, be useful in treating such diseases. It is, however, important to preserve physiological NMDA receptor activity while trying to block its excessive, excitotoxic activity. This can possibly be achieved by uncompetitive antagonists, blocking the receptors ion channel when excessively open.
Uncompetitive NMDA receptor antagonists, or channel blockers, enter the channel of the NMDA receptor after it has been activated and thereby block the flow of ions.MK-801, ketamine, amantadine and memantine are examples of such antagonists, see figure 1. The off-rate of an antagonist from the receptors channel is an important factor as too slow off-rate can interfere with normal function of the receptor and too fast off-rate may give ineffective blockade of an excessively open receptor.
Memantine is an example of an uncompetitive channel blocker of the NMDA receptor, with a relatively rapid off-rate and low affinity. At physiological pH its amine group is positively charged and its receptor antagonism is voltage-dependent. It thereby mimics the physiological function of Mg2+ as channel blocker. Memantine only blocks NMDA receptor associated channels during prolonged activation of the receptor, as it occurs under excitotoxic conditions, by replacing magnesium at the binding site. During normal receptor activity the channels only stay open for several milliseconds and under those circumstances memantine is unable to bind within the channels and therefore doesn't interfere with normal synaptic activity.
Variants
GluN1
There are eight variants of the GluN1 subunit produced by alternative splicing of GRIN1:
- GluN1-1a, GluN1-1b; GluN1-1a is the most abundantly expressed form.
- GluN1-2a, GluN1-2b;
- GluN1-3a, GluN1-3b;
- GluN1-4a, GluN1-4b;
GluN2

While a single GluN2 subunit is found in invertebrate organisms, four distinct isoforms of the GluN2 subunit are expressed in vertebrates and are referred to with the nomenclature GluN2A through GluN2D (encoded by GRIN2A, GRIN2B, GRIN2C, GRIN2D). Strong evidence shows that the genes encoding the GluN2 subunits in vertebrates have undergone at least two rounds of gene duplication. They contain the binding-site for glutamate. More importantly, each GluN2 subunit has a different intracellular C-terminal domain that can interact with different sets of signalling molecules. Unlike GluN1 subunits, GluN2 subunits are expressed differentially across various cell types and developmental timepoints and control the electrophysiological properties of the NMDA receptor. In classic circuits, GluN2B is mainly present in immature neurons and in extrasynaptic locations such as growth cones, and contains the binding-site for the selective inhibitor ifenprodil. However, in pyramidal cell synapses in the newly evolved primate dorsolateral prefrontal cortex, GluN2B are exclusively within the postsynaptic density, and mediate higher cognitive operations such as working memory. This is consistent with the expansion in GluN2B actions and expression across the cortical hierarchy in monkeys and humans and across primate cortex evolution.
GluN2B to GluN2A switch
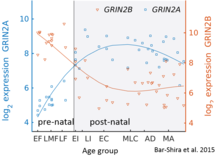
While GluN2B is predominant in the early postnatal brain, the number of GluN2A subunits increases during early development; eventually, GluN2A subunits become more numerous than GluN2B. This is called the GluN2B-GluN2A developmental switch, and is notable because of the different kinetics each GluN2 subunit contributes to receptor function. For instance, greater ratios of the GluN2B subunit leads to NMDA receptors which remain open longer compared to those with more GluN2A. This may in part account for greater memory abilities in the immediate postnatal period compared to late in life, which is the principle behind genetically altered 'doogie mice'. The detailed time course of this switch in the human cerebellum has been estimated using expression microarray and RNA seq and is shown in the figure on the right.
There are three hypothetical models to describe this switch mechanism:
- Increase in synaptic GluN2A along with decrease in GluN2B
- Extrasynaptic displacement of GluN2B away from the synapse with increase in GluN2A
- Increase of GluN2A diluting the number of GluN2B without the decrease of the latter.
The GluN2B and GluN2A subunits also have differential roles in mediating excitotoxic neuronal death. The developmental switch in subunit composition is thought to explain the developmental changes in NMDA neurotoxicity. Homozygous disruption of the gene for GluN2B in mice causes perinatal lethality, whereas disruption of the GluN2A gene produces viable mice, although with impaired hippocampal plasticity. One study suggests that reelin may play a role in the NMDA receptor maturation by increasing the GluN2B subunit mobility.
GluN2B to GluN2C switch
Granule cell precursors (GCPs) of the cerebellum, after undergoing symmetric cell division in the external granule-cell layer (EGL), migrate into the internal granule-cell layer (IGL) where they down-regulate GluN2B and activate GluN2C, a process that is independent of neuregulin beta signaling through ErbB2 and ErbB4 receptors.
Role in excitotoxicity
NMDA receptors have been implicated by a number of studies to be strongly involved with excitotoxicity. Because NMDA receptors play an important role in the health and function of neurons, there has been much discussion on how these receptors can affect both cell survival and cell death. Recent evidence supports the hypothesis that overstimulation of extrasynaptic NMDA receptors has more to do with excitotoxicity than stimulation of their synaptic counterparts. In addition, while stimulation of extrasynaptic NMDA receptors appear to contribute to cell death, there is evidence to suggest that stimulation of synaptic NMDA receptors contributes to the health and longevity of the cell. There is ample evidence to support the dual nature of NMDA receptors based on location, and the hypothesis explaining the two differing mechanisms is known as the "localization hypothesis".
Differing cascade pathways
In order to support the localization hypothesis, it would be necessary to show differing cellular signaling pathways are activated by NMDA receptors based on its location within the cell membrane. Experiments have been designed to stimulate either synaptic or non-synaptic NMDA receptors exclusively. These types of experiments have shown that different pathways are being activated or regulated depending on the location of the signal origin. Many of these pathways use the same protein signals, but are regulated oppositely by NMDARs depending on its location. For example, synaptic NMDA excitation caused a decrease in the intracellular concentration of p38 mitogen-activated protein kinase (p38MAPK). Extrasynaptic stimulation NMDARs regulated p38MAPK in the opposite fashion, causing an increase in intracellular concentration. Experiments of this type have since been repeated with the results indicating these differences stretch across many pathways linked to cell survival and excitotoxicity.
Two specific proteins have been identified as a major pathway responsible for these different cellular responses ERK1/2, and Jacob. ERK1/2 is responsible for phosphorylation of Jacob when excited by synaptic NMDARs. This information is then transported to the nucleus. Phosphorylation of Jacob does not take place with extrasynaptic NMDA stimulation. This allows the transcription factors in the nucleus to respond differently based in the phosphorylation state of Jacob.
Neural plasticity
NMDA receptors (NMDARs) critically influence the induction of synaptic plasticity. NMDARs trigger both long-term potentiation (LTP) and long-term depression (LTD) via fast synaptic transmission. Experimental data suggest that extrasynaptic NMDA receptors inhibit LTP while producing LTD. Inhibition of LTP can be prevented with the introduction of a NMDA antagonist. A theta burst stimulation that usually induces LTP with synaptic NMDARs, when applied selectively to extrasynaptic NMDARs produces a LTD. Experimentation also indicates that extrasynaptic activity is not required for the formation of LTP. In addition, both synaptic and extrasynaptic activity are involved in expressing a full LTD.
Role of differing subunits
Another factor that seems to affect NMDAR induced toxicity is the observed variation in subunit makeup. NMDA receptors are heterotetramers with two GluN1 subunits and two variable subunits. Two of these variable subunits, GluN2A and GluN2B, have been shown to preferentially lead to cell survival and cell death cascades respectively. Although both subunits are found in synaptic and extrasynaptic NMDARs there is some evidence to suggest that the GluN2B subunit occurs more frequently in extrasynaptic receptors. This observation could help explain the dualistic role that NMDA receptors play in excitotoxicity.
Despite the compelling evidence and the relative simplicity of these two theories working in tandem, there is still disagreement about the significance of these claims. Some problems in proving these theories arise with the difficulty of using pharmacological means to determine the subtypes of specific NMDARs. In addition, the theory of subunit variation does not explain how this effect might predominate, as it is widely held that the most common tetramer, made from two GluN1 subunits and one of each subunit GluN2A and GluN2B, makes up a high percentage of the NMDARs.
Excitotoxicity in a clinical setting
Excitotoxicity has been thought to play a role in the degenerative properties of neurodegenerative conditions since the late 1950s. NMDA receptors seem to play an important role in many of these degenerative diseases affecting the brain. Most notably, excitotoxic events involving NMDA receptors have been linked to Alzheimer's disease and Huntington's disease, as well as with other medical conditions such as strokes and epilepsy. Treating these conditions with one of the many known NMDA receptor antagonists, however, leads to a variety of unwanted side effects, some of which can be severe. These side effects are, in part, observed because the NMDA receptors do not just signal for cell death but also play an important role in its vitality. Treatment for these conditions might be found in blocking NMDA receptors not found at the synapse. One class of excitotoxicity in disease includes gain-of-function mutations in GRIN2B and GRIN1 associated with cortical malformations, such as polymicrogyria.
Ligands
Agonists


Activation of NMDA receptors requires binding of glutamate or aspartate (aspartate does not stimulate the receptors as strongly). In addition, NMDARs also require the binding of the co-agonist glycine for the efficient opening of the ion channel, which is a part of this receptor.
D-Serine has also been found to co-agonize the NMDA receptor with even greater potency than glycine. It is produced by serine racemase, and is enriched in the same areas as NMDA receptors. Removal of D-serine can block NMDA-mediated excitatory neurotransmission in many areas. Recently, it has been shown that D-serine can be released both by neurons and astrocytes to regulate NMDA receptors.
NMDA receptor (NMDAR)-mediated currents are directly related to membrane depolarization. NMDA agonists therefore exhibit fast Mg2+ unbinding kinetics, increasing channel open probability with depolarization. This property is fundamental to the role of the NMDA receptor in memory and learning, and it has been suggested that this channel is a biochemical substrate of Hebbian learning, where it can act as a coincidence detector for membrane depolarization and synaptic transmission.
Examples
Some known NMDA receptor agonists include:
-
Amino acids and amino acid derivatives
- Aspartic acid (aspartate) (D-aspartic acid, L-aspartic acid) – endogenous glutamate site agonist. The word N-methyl-D-aspartate (NMDA) is partially derived from D-Aspartate.
-
Glutamic acid (glutamate) – endogenous glutamate site agonist
- Tetrazolylglycine – synthetic glutamate site agonist
- Homocysteic acid – endogenous glutamate site agonist
- Ibotenic acid – naturally occurring glutamate site agonist found in Amanita muscaria
- Quinolinic acid (quinolinate) – endogenous glutamate site agonist
- Glycine – endogenous glycine site agonist
-
Positive allosteric modulators
- Cerebrosterol – endogenous weak positive allosteric modulator
- Cholesterol – endogenous weak positive allosteric modulator
- Dehydroepiandrosterone (DHEA) – endogenous weak positive allosteric modulator
- Dehydroepiandrosterone sulfate (DHEA-S) – endogenous weak positive allosteric modulator
- Nebostinel (neboglamine) – synthetic positive allosteric modulator of the glycine site
- Pregnenolone sulfate – endogenous weak positive allosteric modulator
- Polyamines
- Spermidine – endogenous polyamine site agonist
- Spermine – endogenous polyamine site agonist
Neramexane
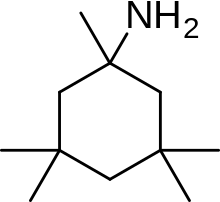
An example of memantine derivative is neramexane which was discovered by studying number of aminoalkyl cyclohexanes, with memantine as the template, as NMDA receptor antagonists. Neramexane binds to the same site as memantine within the NMDA receptor associated channel and with comparable affinity. It does also show very similar bioavailability and blocking kinetics in vivo as memantine. Neramexane went to clinical trials for four indications, including Alzheimer's disease.
Partial agonists

N-Methyl-D-aspartic acid (NMDA), which the NMDA receptor was named after, is a partial agonist of the active or glutamate recognition site.
3,5-Dibromo-L-phenylalanine, a naturally occurring halogenated derivative of L-phenylalanine, is a weak partial NMDA receptor agonist acting on the glycine site. 3,5-Dibromo-L-phenylalanine has been proposed a novel therapeutic drug candidate for treatment of neuropsychiatric disorders and diseases such as schizophrenia, and neurological disorders such as ischemic stroke and epileptic seizures.
Other partial agonists of the NMDA receptor acting on novel sites such as rapastinel (GLYX-13) and apimostinel (NRX-1074) are now viewed for the development of new drugs with antidepressant and analgesic effects without obvious psychotomimetic activities.
Examples
- Aminocyclopropanecarboxylic acid (ACC) – synthetic glycine site partial agonist
- Cycloserine (D-cycloserine) – naturally occurring glycine site partial agonist found in Streptomyces orchidaceus
- HA-966 – synthetic glycine site weak partial agonist
- Homoquinolinic acid – synthetic glutamate site partial agonist
- N-Methyl-D-aspartic acid (NMDA) – synthetic glutamate site partial agonist
Positive allosteric modulators include:
- Zelquistinel (AGN-241751) - synthetic novel site partial agonist
- Apimostinel (NRX-1074) – synthetic novel site partial agonist
- Rapastinel (GLYX-13) – synthetic novel site partial agonist
Antagonists

Antagonists of the NMDA receptor are used as anesthetics for animals and sometimes humans, and are often used as recreational drugs due to their hallucinogenic properties, in addition to their unique effects at elevated dosages such as dissociation. When certain NMDA receptor antagonists are given to rodents in large doses, they can cause a form of brain damage called Olney's lesions. NMDA receptor antagonists that have been shown to induce Olney's lesions include ketamine, phencyclidine, and dextrorphan (a metabolite of dextromethorphan), as well as some NMDA receptor antagonists used only in research environments. So far, the published research on Olney's lesions is inconclusive in its occurrence upon human or monkey brain tissues with respect to an increase in the presence of NMDA receptor antagonists.
Most NMDAR antagonists are uncompetitive or noncompetitive blockers of the channel pore or are antagonists of the glycine co-regulatory site rather than antagonists of the active/glutamate site.
Examples
Common agents in which NMDA receptor antagonism is the primary or a major mechanism of action:
- 4-Chlorokynurenine (AV-101) – glycine site antagonist; prodrug of 7-chlorokynurenic acid
- 7-Chlorokynurenic acid – glycine site antagonist
- Agmatine – endogenous polyamine site antagonist
- Argiotoxin-636 – naturally occurring dizocilpine or related site antagonist found in Argiope venom
- AP5 – glutamate site antagonist
- AP7 – glutamate site antagonist
- CGP-37849 – glutamate site antagonist
- Delucemine (NPS-1506) – dizocilpine or related site antagonist; derived from argiotoxin-636
- Dextromethorphan (DXM) – dizocilpine site antagonist; prodrug of dextrorphan
- Dextrorphan (DXO) – dizocilpine site antagonist
- Dexanabinol – dizocilpine-related site antagonist
- Diethyl ether – unknown site antagonist
- Diphenidine – dizocilpine site antagonist
- Dizocilpine (MK-801) – dizocilpine site antagonist
- Eliprodil – ifenprodil site antagonist
- Esketamine – dizocilpine site antagonist
- Hodgkinsine – undefined site antagonist
- Ifenprodil – ifenprodil site antagonist
- Kaitocephalin – naturally occurring glutamate site antagonist found in Eupenicillium shearii
- Ketamine – dizocilpine site antagonist
- Kynurenic acid – endogenous glycine site antagonist
- Lanicemine – low-trapping dizocilpine site antagonist
- LY-235959 – glutamate site antagonist
- Memantine – low-trapping dizocilpine site antagonist
- Methoxetamine – dizocilpine site antagonist
- Midafotel – glutamate site antagonist
- Nitrous oxide (N2O) – undefined site antagonist
- PEAQX – glutamate site antagonist
- Perzinfotel – glutamate site antagonist
- Phencyclidine (PCP) – dizocilpine site antagonist
- Phenylalanine - a naturally occurring amino acid, glycine site antagonist
- Psychotridine – undefined site antagonist
- Selfotel – glutamate site antagonist
- Tiletamine – dizocilpine site antagonist
- Traxoprodil – ifenprodil site antagonist
- Xenon – unknown site antagonist
Some common agents in which weak NMDA receptor antagonism is a secondary or additional action include:
- Amantadine – an antiviral and antiparkinsonian drug; low-trapping dizocilpine site antagonist
- Atomoxetine – a norepinephrine reuptake inhibitor used to treat ADHD
- Dextropropoxyphene – an opioid analgesic
- Ethanol (alcohol) – a euphoriant, sedative, and anxiolytic used recreationally; unknown site antagonist
- Guaifenesin – an expectorant
- Huperzine A – a naturally occurring acetylcholinesterase inhibitor and potential antidementia agent
- Ibogaine – a naturally occurring hallucinogen and antiaddictive agent
- Ketobemidone – an opioid analgesic
- Methadone – an opioid analgesic
- Minocycline – an antibiotic
- Tramadol – an atypical opioid analgesic and serotonin releasing agent
Nitromemantine
The NMDA receptor is regulated via nitrosylation and aminoadamantane can be used as a target-directed shuttle to bring nitrogen oxide (NO) close to the site within the NMDA receptor where it can nitrosylate and regulate the ion channel conductivity. A NO donor that can be used to decrease NMDA receptor activity is the alkyl nitrate nitroglycerin. Unlike many other NO donors, alkyl nitrates do not have potential NO associated neurotoxic effects. Alkyl nitrates donate NO in the form of a nitro group as seen in figure 7, -NO2-, which is a safe donor that avoids neurotoxicity. The nitro group must be targeted to the NMDA receptor, otherwise other effects of NO such as dilatation of blood vessels and consequent hypotension could result.Nitromemantine is a second-generation derivative of memantine, it reduces excitotoxicity mediated by overactivation of the glutamatergic system by blocking NMDA receptor without sacrificing safety. Provisional studies in animal models show that nitromemantines are more effective than memantine as neuroprotectants, both in vitro and in vivo. Memantine and newer derivatives could become very important weapons in the fight against neuronal damage.

Negative allosteric modulators include:
- 25-Hydroxycholesterol – endogenous weak negative allosteric modulator
- Conantokins – naturally occurring negative allosteric modulators of the polyamine site found in Conus geographus
Modulators
Examples
The NMDA receptor is modulated by a number of endogenous and exogenous compounds:
- Aminoglycosides have been shown to have a similar effect to polyamines, and this may explain their neurotoxic effect.
- CDK5 regulates the amount of NR2B-containing NMDA receptors on the synaptic membrane, thus affecting synaptic plasticity.
- Polyamines do not directly activate NMDA receptors, but instead act to potentiate or inhibit glutamate-mediated responses.
- Reelin modulates NMDA function through Src family kinases and DAB1. significantly enhancing LTP in the hippocampus.
- Src kinase enhances NMDA receptor currents.
- Na+, K+ and Ca2+ not only pass through the NMDA receptor channel but also modulate the activity of NMDA receptors.
- Zn2+ and Cu2+ generally block NMDA current activity in a noncompetitive and a voltage-independent manner. However zinc may potentiate or inhibit the current depending on the neural activity.
- Pb2+ is a potent NMDAR antagonist. Presynaptic deficits resulting from Pb2+ exposure during synaptogenesis are mediated by disruption of NMDAR-dependent BDNF signaling.
- Proteins of the major histocompatibility complex class I are endogenous negative regulators of NMDAR-mediated currents in the adult hippocampus, and are required for appropriate NMDAR-induced changes in AMPAR trafficking and NMDAR-dependent synaptic plasticity and learning and memory.
- The activity of NMDA receptors is also strikingly sensitive to the changes in pH, and partially inhibited by the ambient concentration of H+ under physiological conditions. The level of inhibition by H+ is greatly reduced in receptors containing the NR1a subtype, which contains the positively charged insert Exon 5. The effect of this insert may be mimicked by positively charged polyamines and aminoglycosides, explaining their mode of action.
- NMDA receptor function is also strongly regulated by chemical reduction and oxidation, via the so-called "redox modulatory site." Through this site, reductants dramatically enhance NMDA channel activity, whereas oxidants either reverse the effects of reductants or depress native responses. It is generally believed that NMDA receptors are modulated by endogenous redox agents such as glutathione, lipoic acid, and the essential nutrient pyrroloquinoline quinone.
Development of NMDA receptor antagonists
The main problem with the development of NMDA antagonists for neuroprotection is that physiological NMDA receptor activity is essential for normal neuronal function. Complete blockade of all NMDA receptor activity results in side effects such as hallucinations, agitation and anesthesia. To be clinically relevant, an NMDA receptor antagonist must limit its action to blockade of excessive activation, without limiting normal function of the receptor.
Competitive NMDA receptor antagonists
Competitive NMDA receptor antagonists, which were developed first, are not a good option because they compete and bind to the same site (NR2 subunit) on the receptor as the agonist, glutamate, and therefore block normal function also. They will block healthy areas of the brain prior to having an impact on pathological areas, because healthy areas contain lower levels of agonist than pathological areas. These antagonists can be displaced from the receptor by high concentration of glutamate which can exist under excitotoxic circumstances.
Noncompetitive NMDA receptor antagonists
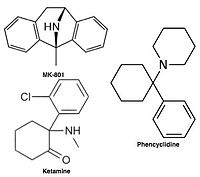
Uncompetitive NMDA receptor antagonists block within the ion channel at the Mg2+ site (pore region) and prevent excessive influx of Ca2+. Noncompetitive antagonism refers to a type of block that an increased concentration of glutamate cannot overcome, and is dependent upon prior activation of the receptor by the agonist, i.e. it only enters the channel when it is opened by agonist.

Because of these adverse side effects of high affinity blockers, the search for clinically successful NMDA receptor antagonists for neurodegenerative diseases continued and focused on developing low affinity blockers. However the affinity could not be too low and dwell time not too short (as seen with Mg2+) where membrane depolarization relieves the block. The discovery was thereby development of uncompetitive antagonist with longer dwell time than Mg2+ in the channel but shorter than MK-801. That way the drug obtained would only block excessively open NMDA receptor associated channels but not normal neurotransmission. Memantine is that drug. It is a derivative of amantadine which was first an anti-influenza agent but was later discovered by coincidence to have efficacy in Parkinson's disease. Chemical structures of memantine and amantadine can be seen in figure 5. The compound was first thought to be dopaminergic or anticholinergic but was later found to be an NMDA receptor antagonist.
Memantine is the first drug approved for treatment of severe and more advanced Alzheimer's disease, which for example anticholinergic drugs do not do much good for. It helps recovery of synaptic function and in that way improves impaired memory and learning. In 2015 memantine is also in trials for therapeutic importance in additional neurological disorders.
Many second-generation memantine derivatives have been in development that may show even better neuroprotective effects, where the main thought is to use other safe but effective modulatory sites on the NMDA receptor in addition to its associated ion channel.
Structure activity relationship (SAR)

Memantine (1-amino-3,5-dimethyladamantane) is an aminoalkyl cyclohexane derivative and an atypical drug compound with non-planar, three dimensional tricyclic structure. Figure 8 shows SAR for aminoalkyl cyclohexane derivative. Memantine has several important features in its structure for its effectiveness:
- Three-ring structure with a bridgehead amine, -NH2
- The -NH2 group is protonated under physiological pH of the body to carry a positive charge, -NH3+
- Two methyl (CH3) side groups which serve to prolong the dwell time and increase stability as well as affinity for the NMDA receptor channel compared with amantadine (1-adamantanamine).
Despite the small structural difference between memantine and amantadine, two adamantane derivatives, the affinity for the binding site of NR1/NR2B subunit is much greater for memantine. In patch-clamp measurements memantine has an IC50 of (2.3+0.3) μM while amantadine has an IC50 of (71.0+11.1) μM. The binding site with the highest affinity is called the dominant binding site. It involves a connection between the amine group of memantine and the NR1-N161 binding pocket of the NR1/NR2B subunit. The methyl side groups play an important role in increasing the affinity to the open NMDA receptor channels and making it a much better neuroprotective drug than amantadine. The binding pockets for the methyl groups are considered to be at the NR1-A645 and NR2B-A644 of the NR1/NR2B. The binding pockets are shown in figure 2. Memantine binds at or near to the Mg2+ site inside the NMDA receptor associated channel. The -NH2 group on memantine, which is protonated under physiological pH of the body, represents the region that binds at or near to the Mg2+ site. Adding two methyl groups to the -N on the memantine structure has shown to decrease affinity, giving an IC50 value of (28.4+1.4) μM.
Second generation derivative of memantine; nitromemantine
Several derivatives of Nitromemantine, a second-generation derivative of memantine, have been synthesized in order to perform a detailed structure activity relationship (SAR) of these novel drugs. One class, containing a nitro (NO2) group opposite to the bridgehead amine (NH2), showed a promising outcome. Nitromemantine utilizes memantine binding site on the NMDA receptor to target the NOx (X= 1 or 2) group for interaction with the S- nitrosylation/redox site external to the memantine binding site. Lengthening the side chains of memantine compensates for the worse drug affinity in the channel associated with the addition of the –ONO2 group
Therapeutic application
Excitotoxicity is implied to be involved in some neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, Huntington's disease and amyotrophic lateral sclerosis. Blocking of NMDA receptors could therefore, in theory, be useful in treating such diseases. It is, however, important to preserve physiological NMDA receptor activity while trying to block its excessive, excitotoxic activity. This can possibly be achieved by uncompetitive antagonists, blocking the receptors ion channel when excessively open
Memantine is an example of uncompetitive NMDA receptor antagonist that has approved indication for the neurodegenerative disease Alzheimer's disease. In 2015 memantine is still in clinical trials for additional neurological diseases.
Receptor modulation
The NMDA receptor is a non-specific cation channel that can allow the passage of Ca2+ and Na+ into the cell and K+ out of the cell. The excitatory postsynaptic potential (EPSP) produced by activation of an NMDA receptor increases the concentration of Ca2+ in the cell. The Ca2+ can in turn function as a second messenger in various signaling pathways. However, the NMDA receptor cation channel is blocked by Mg2+ at resting membrane potential. Magnesium unblock is not instantaneous, to unblock all available channels, the postsynaptic cell must be depolarized for a sufficiently long period of time (in the scale of milliseconds).
Therefore, the NMDA receptor functions as a "molecular coincidence detector". Its ion channel opens only when the following two conditions are met: glutamate is bound to the receptor, and the postsynaptic cell is depolarized (which removes the Mg2+ blocking the channel). This property of the NMDA receptor explains many aspects of long-term potentiation (LTP) and synaptic plasticity.
In a resting-membrane potential, the NMDA receptor pore is opened allowing for an influx of external magnesium ions binding to prevent further ion permeation. External magnesium ions are in a millimolar range while intracellular magnesium ions are at a micromolar range to result in negative membrane potential. NMDA receptors are modulated by a number of endogenous and exogenous compounds and play a key role in a wide range of physiological (e.g., memory) and pathological processes (e.g., excitotoxicity). Magnesium works to potentiate NMDA-induced responses at positive membrane potentials while blocking the NMDA channel. The use of calcium, potassium, and sodium are used to modulate the activity of NMDARs passing through the NMDA membrane. Changes in H+ concentration can partially inhibit the activity of NMDA receptors in different physiological conditions.
Clinical significance
NMDAR antagonists like ketamine, esketamine, tiletamine, phencyclidine, nitrous oxide, and xenon are used as general anesthetics. These and similar drugs like dextromethorphan and methoxetamine also produce dissociative, hallucinogenic, and euphoriant effects and are used as recreational drugs.
NMDAR-targeted compounds, including ketamine, esketamine (JNJ-54135419), rapastinel (GLYX-13), apimostinel (NRX-1074), zelquistinel (AGN-241751), 4-chlorokynurenine (AV-101), and rislenemdaz (CERC-301, MK-0657), are under development for the treatment of mood disorders, including major depressive disorder and treatment-resistant depression. In addition, ketamine is already employed for this purpose as an off-label therapy in some clinics.
Research suggests that tianeptine produces antidepressant effects through indirect alteration and inhibition of glutamate receptor activity and release of BDNF, in turn affecting neural plasticity. Tianeptine also acts on the NMDA and AMPA receptors. In animal models, tianeptine inhibits the pathological stress-induced changes in glutamatergic neurotransmission in the amygdala and hippocampus.
Memantine, a low-trapping NMDAR antagonist, is approved in the United States and Europe for the treatment of moderate-to-severe Alzheimer's disease, and has now received a limited recommendation by the UK's National Institute for Health and Care Excellence for patients who fail other treatment options.
Cochlear NMDARs are the target of intense research to find pharmacological solutions to treat tinnitus. NMDARs are associated with a rare autoimmune disease, anti-NMDA receptor encephalitis (also known as NMDAR encephalitis), that usually occurs due to cross-reactivity of antibodies produced by the immune system against ectopic brain tissues, such as those found in teratoma. These are known as anti-glutamate receptor antibodies.
Compared to dopaminergic stimulants like methamphetamine, the NMDAR antagonist phencyclidine can produce a wider range of symptoms that resemble schizophrenia in healthy volunteers, in what has led to the glutamate hypothesis of schizophrenia. Experiments in which rodents are treated with NMDA receptor antagonist are today the most common model when it comes to testing of novel schizophrenia therapies or exploring the exact mechanism of drugs already approved for treatment of schizophrenia.
NMDAR antagonists, for instance eliprodil, gavestinel, licostinel, and selfotel have been extensively investigated for the treatment of excitotoxicity-mediated neurotoxicity in situations like ischemic stroke and traumatic brain injury, but were unsuccessful in clinical trials used in small doses to avoid sedation, but NMDAR antagonists can block Spreading Depolarizations in animals and in patients with brain injury. This use have not been tested in clinical trials yet.
See also
External links
-
 Media related to NMDA receptor at Wikimedia Commons
Media related to NMDA receptor at Wikimedia Commons - NMDA receptor pharmacology
- Motor Discoordination Results from Combined Gene Disruption of the NMDA Receptor NR2A and NR2C Subunits, But Not from Single Disruption of the NR2A or NR2C Subunit
- A schematic diagram summarizes three potential models for the switching of NR2A and NR2B subunits at developing synapses
- Drosophila NMDA receptor 1 - The Interactive Fly
| Concepts | |
|---|---|
| People | |
| Methods | |
| Systems | |