
| Nodular fasciitis | |
|---|---|
| Other names | Nodular pseudosarcomatous fasciitis, subcutaneous pseudosarcomatous fibromatosis |
 | |
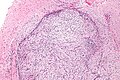
| Micrograph of nodular fasciitis showing the haphazard arrangement of cells (tissue culture-like pattern). H&E stain. | |
| Specialty |
Rheumatology |
| Types | Cranial fasciitis, intravascular fasciitis |
Nodular fasciitis (NF) is a benign, soft tissue tumor composed of myofibroblasts (i.e. immature cells that contain features of myocytes and fibroblasts) that typically occurs in subcutaneous tissue (i.e. lowermost layer of the skin), fascia (i.e. band of connective tissue just beneath the skin), and/or muscles. The literature sometimes titles rare NF variants according to their tissue locations. The most frequently used and important of these are: cranial fasciitis (NF variants that occur in the soft and hard tissues of the skull) and intravascular fasciitis (NF variants that involve arteries and/or veins). In 2020, the World Health Organization classified nodular fasciitis as in the category of benign fibroblastic/myofibroblastic tumors. NF is the most common of the benign fibroblastic proliferative tumors of soft tissue and exceeds in frequency any other tumor or tumor-like lesion in this group of tumors.
Nodular fasciitis is a rapidly growing, usually self-limiting neoplasm that occurs primarily but not exclusively in adults. Due to its rapid growth, NF has often been misdiagnosed as a malignant tumor, usually a sarcoma. Indeed, NF was originally termed subcutaneous pseudosarcomatous fibromatosis when first described in 1955 by Konwaler et al. The correct diagnosis of a tumor as NF is pivotal to prevent its overtreatment as a more aggressive and/or malignant growth.
NF tumors have long been mentioned as local reactions to traumatic injuries in at least some cases. While it still may be precipitated by such injuries, recent studies indicate that NFs are true neoplasms (i.e. abnormal proliferations of cells even after any precipitating event is removed). This is because up to 92% of NF cases have a specific type of fusion gene in their tumor cells. Fusion genes are abnormal and potentially tumor-inducing genes formed by mergers between parts of two different genes. The fusion gene involved in NF merges part of the USP6 gene to a part of any one of numerous other genes. The USP6 gene is responsible for producing ubiquitin carboxyl-terminal hydrolase 6a, a deubiquitinating enzyme that indirectly regulates various signaling pathways which regulate cell growth and death. All of the USP6-containing fusion genes overproduce a chimeric protein (i.e. an abnormal protein consisting of parts derived from different genes) with uncontrolled deubiquitinating enzyme activity that is thought to contribute to the development of NF.
Presentation
Nodular fasciitis occurs in all age groups but more often affects 20-40 year old individuals. Males and females are equally affected. NF tumors, which may be tender or painful, typically present as rapidly growing solitary lesions that reach a final size (usually of 2–3 cm.) within a few weeks. They are located in the upper limbs (39–54% of cases), trunk (15–20% of cases), lower limbs (16–18% of cases), and head or neck area (20% of cases). (Involvement of the head or neck area is more commonly observed in children.) Rare cases off NF have occurred in the: 1) joints of the knee (18 cases), shoulder (3 cases), hand (3 cases), elbow (2 cases), hip (1 case), ankle (1 case), and temporomandibular joint (1 case); or 2) peripheral nerves of the arm (i.e. ulnar nerve, 2 cases) or median nerve, 1 case), hip/lower limb (i.e. sciatic nerve, 1 case), and lumbar spine/pelvis/thigh (i.e. the obturator nerve, 1 case). Individual cases of NF have been reported to occur in the lower female genital tract, bladder, prostate, tongue, and parotid gland. In some cases, NF tumors have regressed after incisional biopsy, i.e. a biopsy that takes a small part of the diseased tissue.
The cranial fasciitis variant of NF occurs in the soft and hard cranial tissues of the outer layers of the skull. Patients with this variant are more commonly males than females and almost exclusively between 3 weeks and 6 years of age. They typically present with a tumor in areas of the head that lay directly over the temporal or parietal bones. Individual cases have been reported to occur in the lower jaw, frontonasal region, anterior nasal spine (i.e. nasal process of the maxilla), and the orbit (i.e. eye socket)-maxilla area. Characteristically, the tumor is rapidly enlarging, non-painful, rarely regressing without treatment, and potentially expanding into the skull's interior.
In a review of 50 cases, the intravascular fasciitis variant of NF occurred in individuals aged 6 months to 66 years (median age 27 years) with male and females being equally affected (52%:48%). Individuals with this variant commonly presented with a blood vessel-localized tumor of the head and neck area (34% of cases), lower extremity (32% of cases), upper extremity (20% of cases), or trunk (14% of cases). The tumor originates in the small blood vessels of the oral mucosa, eyes, lips, cheeks, tongues, and subcutaneous tissue of the extremities (78% of all cases). About 18% of cases involve major veins i.e. 5 cases involved the common femoral veins and one case each involved the femoral vein, common iliac vein, inferior vena cava, and subclavian vein. Cases involving the ascending aorta have also been reported. The presenting symptoms of these tumors were strictly dependent on their locations and impacts on the involved vasculature. Most cases involving superficial sites presented with a small (mean diameter of 1.5 cm), painless, slowly growing mass. However, tumors growing in deep tissues went unnoticed until they became large (e.g. 15 cm.) enough to obstruct blood flow: two cases involving the ascending aorta presented with the signs and symptoms of acute aortic dissection (e.g. severe pain, congestive heart failure, cardiac arrest, fainting , stroke, ischemic peripheral neuropathy, and/or paraplegia) while 9 cases involving large veins presented with acute swelling, pain, and tissue/organ dysfunctions in the areas drained by the involved veins.
Pathology
The microscopic histopathology of hematoxylin and eosin stained nodular fasciitis tumors (see above and three below figures) consists of spindle-shaped myofibroblastic cells (i.e. cells with features of smooth muscle cells and fibroblasts.). These cells are in a myxoid (i.e. more blue or purple compared to normal connective tissue because of excessive uptake of the hematoxylin stain) or a collagenous (high content of collagen fibers) tissue background. The neoplastic myofibroblasts are arranged in whorls and/or short bundles. These cells may show high rates of replicating as judged by their mitotic index but these mitoses are normal in appearance. The tumor tissues often contain red blood cells, lymphocytes and giant osteoclast-like giant cells and may contain sites of bone-like tissue. NF is sometimes classified into three subtypes based on its predominant histopathological pattern: myxoid or reactive (type I), cellular (type II), and fibrous (type III). These patterns appear related to the duration of the lesion with the myxoid variant tending to have the shortest duration and the cellular and fibrous variants tending to have progressively longer durations.Immunohistochemical analyses indicate that the cells in NF usually express smooth muscle actin, muscle specific actin, and vimentin proteins but generally do not express CD34, S-100 protein, desmin, trypsin, factor VIII, F4/80 (also termed macrophage-specific antigen), or HLA-DR1 proteins. Uncommonly, the cells in NF tumors also express the CD68 (a histiocyte-specific marker) protein.

Low magnification
Intermediate magnification

Goldner, 400x



The histopathology and expressions of marker proteins in cranial fasciitis tumors are similar to NF although its tumors tend to be more organized and have higher levels of inflammatory cell infiltrates, vascularity, and involvements of underlying bone than NF.
The histopathology and expressions of marker proteins in intravascular fasciitis tumors are also similar to NF. In comparison to NF, however, its spindle-shaped tumor cells are more often arranged in a storiformed (i.e. whirled) or haphazard pattern and have vesicle-containing nuclei with prominent nucleoli.
Gene abnormalities
Until recently, nodular fasciitis was considered a reaction to trauma or some other unidentified insult at the site where the tumor subsequently developed. However, recent findings indicate that up to 92% of NF tumors involve the self-limiting growth of a clone (i.e. a group of identical cells that share a common ancestry} of neoplastic cells that contain a fusion gene. Fusion genes are abnormal genes consisting of parts from two different genes that form as a result of large scale gene mutations such as chromosomal translocations, interstitial deletions, or inversions. The fusion gene found in NF tumor cells consists of a part of the USP6 gene combined with any one of numerous other genes. Its most common partner gene in NF is the MYH9 (i.e. myosin-9) gene. This USP6-MYH9 fusion gene forms as a result of a translocation of part of the USP6 gene located at band 13.2 on the long (or "q") arm of chromosome 17 with part of the MYH9 gene at band 13.2 on the short (or "p") arm of chromosome 22. Other genes that partner with the USP6 gene to form a fusion gene found in NF include the: RRBP1 (i.e. ribosome binding protein 1) gene, CALU (calumenin) gene, CTNNB1 (catenin beta 1) gene, MIR22HG (produces the MIR22HG long non-coding RNA) gene,SPARC (secreted protein acidic and cysteine rich [also termed osteonectin]) gene, THBS2 (thrombospondin-2) gene, COL6A2 (collagen type VI alpha 2 chain) gene, SEC31A {SEC31 homolog A, COPII coat complex component) gene, EIF5A (eukaryotic translation initiation factor 5A) gene, COL1A1 (collagen type I alpha 1 chain) gene, COL1A2 (collagen type I alpha 2 chain) gene, COL3A1 (collagen type III alpha 1 chain) gene, PAFAH1B1 (platelet activating factor acetylhydrolase 1b regulatory subunit 1) gene, SERPINH1 (serpin family H member 1) gene which produces heat shock protein 47,PDLIM7 (PDZ and LIM domain protein 7) gene, and MYL12A (myosin regulatory light chain 12A) gene. While very few cases have been analyzed to date, USP6-containing fusion genes have been found in the tumor cells of 7 of 15 tested cases of cranial fasciitis and six of six tested cases of intravascular fasciitis.
All of the USP6-containing fusion genes in NF and variants overproduce a chimeric protein containing a part of the USP6 genes product, ubiquitin carboxyl-terminal hydrolase 6a, that has uncontrolled deubiquitinating enzyme activity. This may result in the inappropriate stimulation of multiple cell signaling pathways including the Wnt signaling pathway, one of the JAK-STAT signaling pathways (i.e. the Jak1-STAT3 pathway), the c-Jun signaling pathway, and the NF-κB signaling pathway. Each of these pathways, when inappropriately activated, has been implicated in promoting the development of various tumors and cancers. Further studies are needed to determine which, if any, of these pathways are overactive and can be successfully targeted with specific drug therapies to treat NF and its variants.
Two cases of NF have tumor cells in which the USP6 gene has fuses with the PPP6R3 (i.e. protein phosphatase 6 regulatory subunit 3) gene. In both of these cases, the tumors clearly showed malignant behavior.
Diagnosis
NF may resemble and therefore be misdiagnosed as dermatofibrosarcoma protuberans, fibrosarcoma, malignant fibrous histiocytoma, spindle-cell melanoma (a rare form of melanoma,) leiomyosarcoma, and inflammatory myofibroblastic tumor. The diagnosis of NF and its variants depends on a combination of findings no single one of which is definitive. These findings are: the tumors': presentation (particularly its location in the cranium, within blood vessels, or outside of these areas); histopathology (particularly analyses of tumor cell mitoses which if atypical strongly suggest the tumor is not NF); presence of myofibroblasts that typically express muscle-specific actin, SMA, and vimentin and may express CD68 but generally do not express S100, desmin, trypsin, factor VIII, F4/80, or CD34 (lack of Cd34-expressing cells indicates that the tumor is not a sarcoma); and presence of typical neoplastic cells expressing an USP6-containing fusion gene. (USP6-containing fusion genes also occur in the tumor cells of aneurysmal bone cysts,fibroma of tendon sheaths, giant cell reparative granuloma, and myositis ossificans/fibro-osseous pseudotumor of digits.)
While virtually all cases of NF and variants have had excellent prognoses, the two cases of individuals with an USP6-PPP6R3 fusion gene in their NF tumor cells had less favorable prognoses: their tumors were locally invasive, repeatedly relapsed after surgical removal, and grew progressively larger over 2 and 10 years of observations.
Treatment
Some cases of NF have regressed after being partially biopsied suggesting that a watchful waiting approach may be appropriate after biopsy in some cases of these tumors. In all events, the most common and generally accepted first-line treatment for most cases of NF, craniofacial fasciitis, and intravascular fasciitis tumors is surgical removal. In almost all cases this removal is curative, i.e. tumor recurrences are uncommon, even in cases where the tumor is only partially removed. Furthermore, tumor recurrences are typically cured by simple re-excisions. NF cases have also been successfully treated with corticosteroid drugs (e.g. triamcinolone) injected directly into the tumor; This treatment has been most often used for patients with recurrent tumors. Some studies, however, have suggested reevaluating the diagnosis in recurrent NF tumors to ensure the diagnosis is correct. Surgical and corticosteroid interventions may need to be performed promptly in order to avoid expansion of a cranial tumor into the skull's interior or an intravascular fasciitis tumor from compromising blood flood flow.
Prognosis
Two cases of NF in which their tumor cells expressed a USP6-PPP6R3 fusion gene had highly aggressive, locally invasive, repeatedly recurrent (after surgical removal), and progressively enlarging tumors over 2 and ten years. One case developed multiple metastases (i.e. spread of a tumor to different parts of the body) that along with the primary tumor site were partially controlled with radiation therapy. The other case was treated by wide surgical resection of the tumor; this case did not have metastatic disease and one year after the wide resection had no recurrences.