
| Primary pigmented nodular adrenocortical disease | |
|---|---|
| Other names | PPNAD |
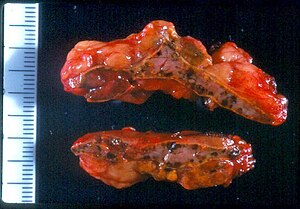 | |
| Macroscopic appearance of adrenal gland in PPNAD following adrenalectomy | |
Primary pigmented nodular adrenocortical disease (PPNAD) was first coined in 1984 by Carney et al. it often occurs in association with Carney complex (CNC). CNC is a rare syndrome that involves the formation of abnormal tumours that cause endocrine hyperactivity.
PPNAD arises due to the enlargement of the cortex of the adrenal glands, resulting in Cushing's syndrome that is independent of the pituitary hormone ACTH.
Signs and symptoms

PPNAD is a rare cause of high cortisol levels in the blood and often manifests as ACTH-independent Cushing's syndrome. The effects of PPNAD can often be cyclical so the symptoms of Cushing's syndrome will not always be as severe, which may complicate diagnosis. The classic symptoms of Cushing's syndrome include rapid central weight gain, a puffy red face and a buffalo hump at the back of the neck due to fat deposits. Skin changes in Cushing's syndrome include thinning and bruising easily, developing striae and hyperpigmentation at skin folds. The hormonal changes can lead to hirsutism, males developing breast tissue, females no longer having periods and both sexes may become infertile. High cortisol levels can lead to psychological disturbances such as anxiety or depression and insomnia. Bone health can deteriorate, leading to an increased fracture risk in people with Cushing's syndrome. PPNAD is unique as it often causes Cushing's at a young age, in children and adolescents. In addition to the other symptoms of Cushing's syndrome, the patient may have a short stature due to interrupted growth because of ACTH suppression.
In 90% of people with PPNAD it is associated with Carney Complex. Carney Complex is usually inherited, however it can also occur sporadically. A visible sign of Carney complex is abnormal skin hyperpigmentation. There may also be myxomas which can appear as lumps in the skin and breast as well as often being present in the heart, which can lead to multiple cardiovascular problems. The majority of people with PPNAD will have some of these signs/symptoms due to the strong association between PPNAD and Carney Complex.
Causes
PPNAD, the endocrine manifestation that comes from Carney Complex (CNC), can be syndromic or isolated. The main cause of isolated PPNAD is a mutation of PRKAR1α, located at 17q22-24, which is the gene encoding the regulatory R1α subunit of protein kinase A. Germline heterozygous PRKAR1α inactivation mutations are present in 80% of CNC patients affected by Cushing's syndrome. There are over 117 mutations of the PRKAR1α gene that can cause CNC, with many of these mutations producing premature stop codons, thus resulting in the complete loss of PRKAR1α protein. CNC patients have also been discovered with an unusually shortened PRKAR1α protein, detected in tumours and leukocytes, following a splice-site mutation, which causes exon-6 skipping. Therefore, both haploinsufficiency and the complete loss of PRKAR1α can lead to the increased PKA activity observed in PPNAD patients, due to the disruption of the cAMP signalling pathway.
Sahut-Barnola et al. used a mouse model to cre-lox knockout the Prkar1a gene specifically from cells of the adrenal cortex and observed that the mice subsequently developed Cushing syndrome that is independent of the pituitary. They also observed that the mutation caused increased PKA activity.
The R1α loss caused the adult adrenal gland became hyperactive and hyperplastic on both sides, as seemingly the foetal adrenal cells within it were not maintained and thus expanded. This established tumoral growths. This mouse KO model phenocopies what happens in human cases of PPNAD.
Inactivation of PDE11A4, located at 2q31-5, has also been identified in PPNAD patients without PRKAR1α mutations. PDE11A4 is the gene encoding phosphodiesterase 11A4, another participant of the cAMP signalling pathway.
Diagnosis
Diagnosis usually occurs upon investigation of a cause for already suspected Cushing's syndrome. High levels of cortisol observed in patients with PPNAD are not suppressed upon administration of dexamethasone (dexamethasone suppression test), and upon MRI or CT imaging, the pituitary will show no abnormalities. Measuring ACTH will confirm that the cause of the patients Cushing's syndrome is ACTH independent. The nature of Cushing's syndrome itself is periodic, which can make diagnosing PPNAD increasingly difficult.
Diagnosis of PPNAD can be difficult to determine preoperatively as CT scan findings can be variable i.e. appear normal or suggest unilateral adrenal lesions therefore impeding the correct diagnosis. NP-59 scintigraphy may be particularly useful in identifying the bilateral nature of the disease.
Gene studies are not necessary for diagnosis as there are clear gross and histological diagnostic markers, as the nodules can usually be seen clearly in both cases A positive family history of PPNAD has been shown to be associated with abnormal histological findings, e.g. mitotic figures, which may further hinder diagnosis. At the point where abdominal CT scanning and pituitary fossa MRI show no clear abnormalities, adrenalectomy may be performed.
Treatment
After diagnosis, it is important for patients to be continually monitored. The most common treatment for PPNAD is bilateral laparoscopic adrenalectomy; the process by which both adrenal glands are removed by a small incision. Patients who have received this treatment will be prescribed mineralocorticoid and glucocorticoid steroids as they are no longer being naturally produced. This is a treatment which has been used and refined since 1984.