
| Serpin (serine protease inhibitor) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
 A serpin (white) with its 'reactive centre loop' (blue) bound to a protease (grey). Once the protease attempts catalysis it will be irreversibly inhibited. (PDB: 1K9O)
| |||||||||||
| Identifiers | |||||||||||
| Symbol | Serpin, SERPIN (root symbol of family) | ||||||||||
| Pfam | PF00079 | ||||||||||
| InterPro | IPR000215 | ||||||||||
| PROSITE | PDOC00256 | ||||||||||
| SCOP2 | 1hle / SCOPe / SUPFAM | ||||||||||
| CDD | cd00172 | ||||||||||
| |||||||||||
Serpins are a superfamily of proteins with similar structures that were first identified for their protease inhibition activity and are found in all kingdoms of life. The acronym serpin was originally coined because the first serpins to be identified act on chymotrypsin-like serine proteases (serine protease inhibitors). They are notable for their unusual mechanism of action, in which they irreversibly inhibit their target protease by undergoing a large conformational change to disrupt the target's active site. This contrasts with the more common competitive mechanism for protease inhibitors that bind to and block access to the protease active site.
Protease inhibition by serpins controls an array of biological processes, including coagulation and inflammation, and consequently these proteins are the target of medical research. Their unique conformational change also makes them of interest to the structural biology and protein folding research communities. The conformational-change mechanism confers certain advantages, but it also has drawbacks: serpins are vulnerable to mutations that can result in serpinopathies such as protein misfolding and the formation of inactive long-chain polymers. Serpin polymerisation not only reduces the amount of active inhibitor, but also leads to accumulation of the polymers, causing cell death and organ failure.
Although most serpins control proteolytic cascades, some proteins with a serpin structure are not enzyme inhibitors, but instead perform diverse functions such as storage (as in egg white—ovalbumin), transport as in hormone carriage proteins (thyroxine-binding globulin, cortisol-binding globulin) and molecular chaperoning (HSP47). The term serpin is used to describe these members as well, despite their non-inhibitory function, since they are evolutionarily related.
History
Protease inhibitory activity in blood plasma was first reported in the late 1800s, but it was not until the 1950s that the serpins antithrombin and alpha 1-antitrypsin were isolated, with the subsequent recognition of their close family homology in 1979. That they belonged to a new protein family became apparent on their further alignment with the non-inhibitory egg-white protein ovalbumin, to give what was initially called the alpha1-antitrypsin-antithrombin III-ovalbumin superfamily of serine proteinase inhibitors, but was subsequently succinctly renamed as the Serpins. The initial characterisation of the new family centred on alpha1-antitrypsin, a serpin present in high concentration in blood plasma, the common genetic disorder of which was shown to cause a predisposition to the lung disease emphysema and to liver cirrhosis. The identification of the S and Z mutations responsible for the genetic deficiency and the subsequent sequence alignments of alpha1-antitrypsin and antithrombin in 1982 led to the recognition of the close homologies of the active sites of the two proteins, centred on a methionine in alpha1-antitrypsin as an inhibitor of tissue elastase and on arginine in antithrombin as an inhibitor of thrombin.
The critical role of the active centre residue in determining the specificity of inhibition of serpins was unequivocally confirmed by the finding that a natural mutation of the active centre methionine in alpha1-antitrypsin to an arginine, as in antithrombin, resulted in a severe bleeding disorder. This active-centre specificity of inhibition was also evident in the many other families of protease inhibitors but the serpins differed from them in being much larger proteins and also in possessing what was soon apparent as an inherent ability to undergo a change in shape. The nature of this conformational change was revealed with the determination in 1984 of the first crystal structure of a serpin, that of post-cleavage alpha1-antitrypsin. This together with the subsequent solving of the structure of native (uncleaved) ovalbumin indicated that the inhibitory mechanism of the serpins involved a remarkable conformational shift, with the movement of the exposed peptide loop containing the reactive site and its incorporation as a middle strand in the main beta-pleated sheet that characterises the serpin molecule. Early evidence of the essential role of this loop movement in the inhibitory mechanism came from the finding that even minor aberrations in the amino acid residues that form the hinge of the movement in antithrombin resulted in thrombotic disease. Ultimate confirmation of the linked displacement of the target protease by this loop movement was provided in 2000 by the structure of the post-inhibitory complex of alpha1-antitrypsin with trypsin, showing how the displacement results in the deformation and inactivation of the attached protease. Subsequent structural studies have revealed an additional advantage of the conformational mechanism in allowing the subtle modulation of inhibitory activity, as notably seen at tissue level with the functionally diverse serpins in human plasma.
Over 1000 serpins have now been identified, including 36 human proteins, as well as molecules in all kingdoms of life—animals, plants, fungi, bacteria, and archaea—and some viruses. The central feature of all is a tightly conserved framework, which allows the precise alignment of their key structural and functional components based on the template structure of alpha1-antitrypsin. In the 2000s, a systematic nomenclature was introduced in order to categorise members of the serpin superfamily based on their evolutionary relationships. Serpins are therefore the largest and most diverse superfamily of protease inhibitors.
Activity

Most serpins are protease inhibitors, targeting extracellular, chymotrypsin-like serine proteases. These proteases possess a nucleophilic serine residue in a catalytic triad in their active site. Examples include thrombin, trypsin, and human neutrophil elastase. Serpins act as irreversible, suicide inhibitors by trapping an intermediate of the protease's catalytic mechanism.
Some serpins inhibit other protease classes, typically cysteine proteases, and are termed "cross-class inhibitors". These enzymes differ from serine proteases in that they use a nucleophilic cysteine residue, rather than a serine, in their active site. Nonetheless, the enzymatic chemistry is similar, and the mechanism of inhibition by serpins is the same for both classes of protease. Examples of cross-class inhibitory serpins include serpin B4 a squamous cell carcinoma antigen 1 (SCCA-1) and the avian serpin myeloid and erythroid nuclear termination stage-specific protein (MENT), which both inhibit papain-like cysteine proteases.
Biological function and localization
Protease inhibition
Approximately two-thirds of human serpins perform extracellular roles, inhibiting proteases in the bloodstream in order to modulate their activities. For example, extracellular serpins regulate the proteolytic cascades central to blood clotting (antithrombin), the inflammatory and immune responses (antitrypsin, antichymotrypsin, and C1-inhibitor) and tissue remodelling (PAI-1). By inhibiting signalling cascade proteases, they can also affect development. The table of human serpins (below) provides examples of the range of functions performed by human serpin, as well as some of the diseases that result from serpin deficiency.
The protease targets of intracellular inhibitory serpins have been difficult to identify, since many of these molecules appear to perform overlapping roles. Further, many human serpins lack precise functional equivalents in model organisms such as the mouse. Nevertheless, an important function of intracellular serpins may be to protect against the inappropriate activity of proteases inside the cell. For example, one of the best-characterised human intracellular serpins is Serpin B9, which inhibits the cytotoxic granule protease granzyme B. In doing so, Serpin B9 may protect against inadvertent release of granzyme B and premature or unwanted activation of cell death pathways.
Some viruses use serpins to disrupt protease functions in their host. The cowpox viral serpin CrmA (cytokine response modifier A) is used in order to avoid inflammatory and apoptotic responses of infected host cells. CrmA increases infectivity by suppressing its host's inflammatory response through inhibition of IL-1 and IL-18 processing by the cysteine protease caspase-1. In eukaryotes, a plant serpin inhibits both metacaspases and a papain-like cysteine protease.
Non-inhibitory roles
Non-inhibitory extracellular serpins also perform a wide array of important roles. Thyroxine-binding globulin and transcortin transport the hormones thyroxine and cortisol, respectively. The non-inhibitory serpin ovalbumin is the most abundant protein in egg white. Its exact function is unknown, but it is thought to be a storage protein for the developing foetus.Heat shock serpin 47 is a chaperone, essential for proper folding of collagen. It acts by stabilising collagen's triple helix whilst it is being processed in the endoplasmic reticulum.
Some serpins are both protease inhibitors and perform additional roles. For example, the nuclear cysteine protease inhibitor MENT, in birds also acts as a chromatin remodelling molecule in a bird's red blood cells.
Structure
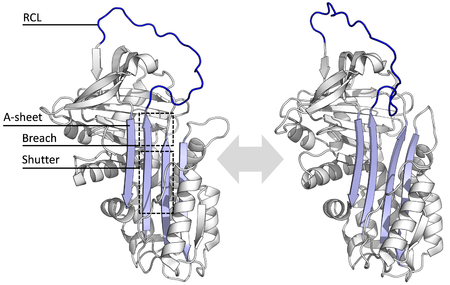
All serpins share a common structure (or fold), despite their varied functions. All typically have three β-sheets (named A, B and C) and eight or nine α-helices (named hA–hI). The most significant regions to serpin function are the A-sheet and the reactive centre loop (RCL). The A-sheet includes two β-strands that are in a parallel orientation with a region between them called the 'shutter', and upper region called the 'breach'. The RCL forms the initial interaction with the target protease in inhibitory molecules. Structures have been solved showing the RCL either fully exposed or partially inserted into the A-sheet, and serpins are thought to be in dynamic equilibrium between these two states. The RCL also only makes temporary interactions with the rest of the structure, and is therefore highly flexible and exposed to the solvent.
The serpin structures that have been determined cover several different conformations, which has been necessary for the understanding of their multiple-step mechanism of action. Structural biology has therefore played a central role in the understanding of serpin function and biology.
Conformational change and inhibitory mechanism
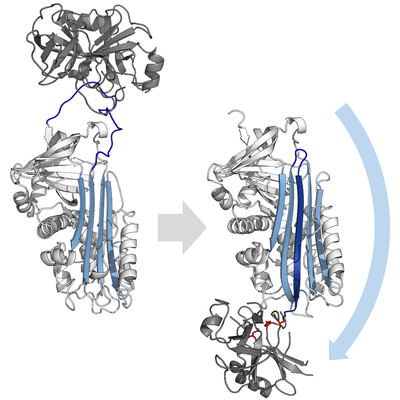
Inhibitory serpins do not inhibit their target proteases by the typical competitive (lock-and-key) mechanism used by most small protease inhibitors (e.g. Kunitz-type inhibitors). Instead, serpins use an unusual conformational change, which disrupts the structure of the protease and prevents it from completing catalysis. The conformational change involves the RCL moving to the opposite end of the protein and inserting into β-sheet A, forming an extra antiparallel β-strand. This converts the serpin from a stressed state, to a lower-energy relaxed state (S to R transition).
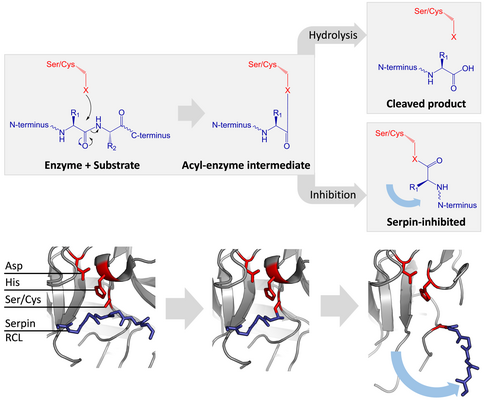
Serine and cysteine proteases catalyse peptide bond cleavage by a two-step process. Initially, the catalytic residue of the active site triad performs a nucleophilic attack on the peptide bond of the substrate. This releases the new N-terminus and forms a covalent ester-bond between the enzyme and the substrate. This covalent complex between enzyme and substrate is called an acyl-enzyme intermediate. For standard substrates, the ester bond is hydrolysed and the new C-terminus is released to complete catalysis. However, when a serpin is cleaved by a protease, it rapidly undergoes the S to R transition before the acyl-enzyme intermediate is hydrolysed. The efficiency of inhibition depends on fact that the relative kinetic rate of the conformational change is several orders of magnitude faster than hydrolysis by the protease.
Since the RCL is still covalently attached to the protease via the ester bond, the S to R transition pulls protease from the top to the bottom of the serpin and distorts the catalytic triad. The distorted protease can only hydrolyse the acyl enzyme intermediate extremely slowly and so the protease remains covalently attached for days to weeks. Serpins are classed as irreversible inhibitors and as suicide inhibitors since each serpin protein permanently inactivates a single protease, and can only function once.
Allosteric activation

The conformational mobility of serpins provides a key advantage over static lock-and-key protease inhibitors. In particular, the function of inhibitory serpins can be regulated by allosteric interactions with specific cofactors. The X-ray crystal structures of antithrombin, heparin cofactor II, MENT and murine antichymotrypsin reveal that these serpins adopt a conformation wherein the first two amino acids of the RCL are inserted into the top of the A β-sheet. The partially inserted conformation is important because co-factors are able to conformationally switch certain partially inserted serpins into a fully expelled form. This conformational rearrangement makes the serpin a more effective inhibitor.
The archetypal example of this situation is antithrombin, which circulates in plasma in a partially inserted relatively inactive state. The primary specificity determining residue (the P1 arginine) points toward the body of the serpin and is unavailable to the protease. Upon binding a high-affinity pentasaccharide sequence within long-chain heparin, antithrombin undergoes a conformational change, RCL expulsion, and exposure of the P1 arginine. The heparin pentasaccharide-bound form of antithrombin is, thus, a more effective inhibitor of thrombin and factor Xa. Furthermore, both of these coagulation proteases also contain binding sites (called exosites) for heparin. Heparin, therefore, also acts as a template for binding of both protease and serpin, further dramatically accelerating the interaction between the two parties. After the initial interaction, the final serpin complex is formed and the heparin moiety is released. This interaction is physiologically important. For example, after injury to the blood vessel wall, heparin is exposed, and antithrombin is activated to control the clotting response. Understanding of the molecular basis of this interaction enabled the development of Fondaparinux, a synthetic form of Heparin pentasaccharide used as an anti-clotting drug.
Latent conformation

Certain serpins spontaneously undergo the S to R transition without having been cleaved by a protease, to form a conformation termed the latent state. Latent serpins are unable to interact with proteases and so are no longer protease inhibitors. The conformational change to latency is not exactly the same as the S to R transition of a cleaved serpin. Since the RCL is still intact, the first strand of the C-sheet has to peel off to allow full RCL insertion.
Regulation of the latency transition can act as a control mechanism in some serpins, such as PAI-1. Although PAI-1 is produced in the inhibitory S conformation, it "auto-inactivates" by changing to the latent state unless it is bound to the cofactor vitronectin. Similarly, antithrombin can also spontaneously convert to the latent state, as an additional modulation mechanism to its allosteric activation by heparin. Finally, the N-terminus of tengpin, a serpin from Thermoanaerobacter tengcongensis, is required to lock the molecule in the native inhibitory state. Disruption of interactions made by the N-terminal region results in spontaneous conformational change of this serpin to the latent conformation.
Conformational change in non-inhibitory functions
Certain non-inhibitory serpins also use the serpin conformational change as part of their function. For example, the native (S) form of thyroxine-binding globulin has high affinity for thyroxine, whereas the cleaved (R) form has low affinity. Similarly, transcortin has higher affinity for cortisol when in its native (S) state, than its cleaved (R) state. Thus, in these serpins, RCL cleavage and the S to R transition has been commandeered to allow for ligand release, rather than protease inhibition.
In some serpins, the S to R transition can activate cell signalling events. In these cases, a serpin that has formed a complex with its target protease, is then recognised by a receptor. The binding event then leads to downstream signalling by the receptor. The S to R transition is therefore used to alert cells to the presence of protease activity. This differs from the usual mechanism whereby serpins affect signalling simply by inhibiting proteases involved in a signalling cascade.
Degradation
When a serpin inhibits a target protease, it forms a permanent complex, which needs to be disposed of. For extracellular serpins, the final serpin-enzyme complexes are rapidly cleared from circulation. One mechanism by which this occurs in mammals is via the low-density lipoprotein receptor-related protein (LRP), which binds to inhibitory complexes made by antithrombin, PA1-1, and neuroserpin, causing cellular uptake. Similarly, the Drosophila serpin, necrotic, is degraded in the lysosome after being trafficked into the cell by the Lipophorin Receptor-1 (homologous to the mammalian LDL receptor family).
Disease and serpinopathies
Serpins are involved in a wide array of physiological functions, and so mutations in genes encoding them can cause a range of diseases. Mutations that change the activity, specificity or aggregation properties of serpins all affect how they function. The majority of serpin-related diseases are the result of serpin polymerisation into aggregates, though several other types of disease-linked mutations also occur. The disorder alpha-1 antitrypsin deficiency is one of the most common hereditary diseases.
Inactivity or absence
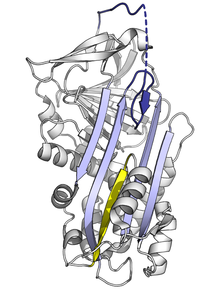
Since the stressed serpin fold is high-energy, mutations can cause them to incorrectly change into their lower-energy conformations (e.g. relaxed or latent) before they have correctly performed their inhibitory role.
Mutations that affect the rate or the extent of RCL insertion into the A-sheet can cause the serpin to undergo its S to R conformational change before having engaged a protease. Since a serpin can only make this conformational change once, the resulting misfired serpin is inactive and unable to properly control its target protease. Similarly, mutations that promote inappropriate transition to the monomeric latent state cause disease by reducing the amount of active inhibitory serpin. For example, the disease-linked antithrombin variants wibble and wobble, both promote formation of the latent state.
The structure of the disease-linked mutant of antichymotrypsin (L55P) revealed another, inactive "δ-conformation". In the δ-conformation, four residues of the RCL are inserted into the top of β-sheet A. The bottom half of the sheet is filled as a result of one of the α-helices (the F-helix) partially switching to a β-strand conformation, completing the β-sheet hydrogen bonding. It is unclear whether other serpins can adopt this conformer, and whether this conformation has a functional role, but it is speculated that the δ-conformation may be adopted by Thyroxine-binding globulin during thyroxine release. The non-inhibitory proteins related to serpins can also cause diseases when mutated. For example, mutations in SERPINF1 cause osteogenesis imperfecta type VI in humans.
In the absence of a required serpin, the protease that it normally would regulate is over-active, leading to pathologies. Consequently, simple deficiency of a serpin (e.g. a null mutation) can result in disease.Gene knockouts, particularly in mice, are used experimentally to determine the normal functions of serpins by the effect of their absence.
Specificity change
In some rare cases, a single amino acid change in a serpin's RCL alters its specificity to target the wrong protease. For example, the Antitrypsin-Pittsburgh mutation (M358R) causes the α1-antitrypsin serpin to inhibit thrombin, causing a bleeding disorder.
Polymerisation and aggregation


The majority of serpin diseases are due to protein aggregation and are termed "serpinopathies". Serpins are vulnerable to disease-causing mutations that promote formation of misfolded polymers due to their inherently unstable structures. Well-characterised serpinopathies include α1-antitrypsin deficiency (alpha-1), which may cause familial emphysema, and sometimes liver cirrhosis, certain familial forms of thrombosis related to antithrombin deficiency, types 1 and 2 hereditary angioedema (HAE) related to deficiency of C1-inhibitor, and familial encephalopathy with neuroserpin inclusion bodies (FENIB; a rare type of dementia caused by neuroserpin polymerisation).
Each monomer of the serpin aggregate exists in the inactive, relaxed conformation (with the RCL inserted into the A-sheet). The polymers are therefore hyperstable to temperature and unable to inhibit proteases. Serpinopathies therefore cause pathologies similarly to other proteopathies (e.g. prion diseases) via two main mechanisms. First, the lack of active serpin results in uncontrolled protease activity and tissue destruction. Second, the hyperstable polymers themselves clog up the endoplasmic reticulum of cells that synthesize serpins, eventually resulting in cell death and tissue damage. In the case of antitrypsin deficiency, antitrypsin polymers cause the death of liver cells, sometimes resulting in liver damage and cirrhosis. Within the cell, serpin polymers are slowly removed via degradation in the endoplasmic reticulum. However, the details of how serpin polymers cause cell death remains to be fully understood.
Physiological serpin polymers are thought to form via domain swapping events, where a segment of one serpin protein inserts into another. Domain-swaps occur when mutations or environmental factors interfere with the final stages of serpin folding to the native state, causing high-energy intermediates to misfold. Both dimer and trimer domain-swap structures have been solved. In the dimer (of antithrombin), the RCL and part of the A-sheet incorporates into the A-sheet of another serpin molecule. The domain-swapped trimer (of antitrypsin) forms via the exchange of an entirely different region of the structure, the B-sheet (with each molecule's RCL inserted into its own A-sheet). It has also been proposed that serpins may form domain-swaps by inserting the RCL of one protein into the A-sheet of another (A-sheet polymerisation). These domain-swapped dimer and trimer structures are thought to be the building blocks of the disease-causing polymer aggregates, but the exact mechanism is still unclear.
Therapeutic strategies
Several therapeutic approaches are in use or under investigation to treat the most common serpinopathy: antitrypsin deficiency. Antitrypsin augmentation therapy is approved for severe antitrypsin deficiency-related emphysema. In this therapy, antitrypsin is purified from the plasma of blood donors and administered intravenously (first marketed as Prolastin). To treat severe antitrypsin deficiency-related disease, lung and liver transplantation has proven effective. In animal models, gene targeting in induced pluripotent stem cells has been successfully used to correct an antitrypsin polymerisation defect and to restore the ability of the mammalian liver to secrete active antitrypsin. Small molecules have also been developed that block antitrypsin polymerisation in vitro.
Evolution
Serpins are the most widely distributed and largest superfamily of protease inhibitors. They were initially believed to be restricted to eukaryote organisms, but have since been found in bacteria, archaea and some viruses. It remains unclear whether prokaryote genes are the descendants of an ancestral prokaryotic serpin or the product of horizontal gene transfer from eukaryotes. Most intracellular serpins belong to a single phylogenetic clade, whether they come from plants or animals, indicating that the intracellular and extracellular serpins may have diverged before the plants and animals. Exceptions include the intracellular heat shock serpin HSP47, which is a chaperone essential for proper folding of collagen, and cycles between the cis-Golgi and the endoplasmic reticulum.
Protease-inhibition is thought to be the ancestral function, with non-inhibitory members the results of evolutionary neofunctionalisation of the structure. The S to R conformational change has also been adapted by some binding serpins to regulate affinity for their targets.
Distribution
Animal
Human
The human genome encodes 16 serpin clades, termed serpinA through serpinP, including 29 inhibitory and 7 non-inhibitory serpin proteins. The human serpin naming system is based upon a phylogenetic analysis of approximately 500 serpins from 2001, with proteins named serpinXY, where X is the clade of the protein and Y the number of the protein within that clade. The functions of human serpins have been determined by a combination of biochemical studies, human genetic disorders, and knockout mouse models.
| Gene name | Common Name | Localisation | Function / Activity | Effect of deficiency | Human disease | Chromosomal location | Protein structure |
|---|---|---|---|---|---|---|---|
| SERPINA1 | α1-antitrypsin | Extracellular | Inhibitor of human neutrophil elastase. The C-terminal fragment of cleaved SERPINA1 may inhibit HIV-1 infection. | Deficiency results in emphysema, polymerisation results in cirrhosis (serpinopathy). | 14q32.1 | 1QLP, 7API, 1D5S | |
| SERPINA2 | Antitrypsin-related protein | Extracellular | Possible pseudogene. | 14q32.1 | |||
| SERPINA3 | α1-antichymotrypsin | Extracellular | Inhibitor of cathepsin G. Additional roles in chromatin condensation in hepatic cells. | Mis-regulation results in Alzheimer's disease (serpinopathy). | 14q32.1 | 1YXA, 2ACH | |
| SERPINA4 | Kallistatin | Extracellular | Inhibitor of kallikrein, regulator of vascular function. | Depletion in hypertensive rats exacerbates renal and cardiovascular injury. | 14q32.1 | ||
| SERPINA5 | Protein C inhibitor | Extracellular | Inhibitor of active protein C. Intracellular role in preventing phagocytosis of bacteria. | Knockout in male mice causes infertility. Accumulation occurs in chronic active plaques in multiple sclerosis. | 14q32.1 | 2OL2, 3B9F | |
| SERPINA6 | Transcortin | Extracellular | Non-inhibitory. Cortisol binding. | Deficiency associated with chronic fatigue. | 14q32.1 | 2V6D, 2VDX, 2VDY | |
| SERPINA7 | Thyroxine-binding globulin | Extracellular | Non-inhibitory. Thyroxine binding. | Deficiency causes hypothyroidism. | Xq22.2 | 2CEO, 2RIV, 2RIW | |
| SERPINA8 | Angiotensinogen | Extracellular | Non-inhibitory, cleavage by renin results in release of angiotensin I. | Knockout in mice causes hypotension. | Variants linked to hypertension. | 1q42-q43 | 2X0B, 2WXW, 2WXX, 2WXY, 2WXZ, 2WY0, 2WY1 |
| SERPINA9 | Centerin / GCET1 | Extracellular | Inhibitory, maintenance of naive B cells. | Strongly expressed in most B-cell lymphomas. | 14q32.1 | ||
| SERPINA10 | Protein Z-related protease inhibitor | Extracellular | Binds protein Z and inactivates factor Xa and factor XIa. | 14q32.1 | 3F1S, 3H5C | ||
| SERPINA11 | – | Probably extracellular | Unknown | 14q32.13 | |||
| SERPINA12 | Vaspin | Extracellular | Inhibitor of Kallikrein-7. Insulin-sensitizing adipocytokine. | High plasma levels associated with type II diabetes. | 14q32.1 | 4IF8 | |
| SERPINA13 | – | Probably extracellular | Unknown | 14q32 | |||
| SERPINB1 | Monocyte neutrophil elastase inhibitor | Intracellular | Inhibitor of neutrophil elastase. | Knockout in mice causes neutrophil survival defect and immune deficiency. | 6p25 | 1HLE | |
| SERPINB2 | Plasminogen activator inhibitor-2 | Intracellular/extracellular | Inhibitor of extracellular uPA. Intracellular function unclear, but may protect against viral infection. | Deficiency in mice reduces immune response to nematode infection. Knockout in mice causes no obvious phenotype. | 18q21.3 | 1BY7 | |
| SERPINB3 | Squamous cell carcinoma antigen-1 (SCCA-1) | Intracellular | Inhibitor of papain-like cysteine proteases and cathepsins K, L and S. | Knockout in mice of Serpinb3a (the murine homolog of both human SERPINB3 and SERPINB4) have reduced mucus production in a murine model of asthma. | 18q21.3 | 2ZV6 | |
| SERPINB4 | Squamous cell carcinoma antigen-2 (SCCA-2) | Intracellular | Inhibitor of chymotrypsin-like serine proteases, cathepsin G and chymase. | Knockout in mice of Serpinb3a (the murine homolog of both human SERPINB3 and SERPINB4) have reduced mucus production in a murine model of asthma. | 18q21.3 | ||
| SERPINB5 | Maspin | Intracellular | Non-inhibitory, function unclear (see also maspin) | Knockout in mice originally reported as lethal, but subsequently shown to have no obvious phenotype. Expression may be a prognostic indicator that reflects expression of a neighbouring tumour suppressor gene (the phosphatase PHLPP1). | 18q21.3 | 1WZ9 | |
| SERPINB6 | PI-6 | Intracellular | Inhibitor of cathepsin G. | Knockout in mice causes hearing loss and mild neutropenia. | Deficiency associated with hearing loss. | 6p25 | |
| SERPINB7 | Megsin | Intracellular | Involved in megakaryocyte maturation. | Over-expression in mice causes kidney disease. Knockout in mice does not cause histological abnormalities. | Mutations associated with Nagashima-type Palmoplantar Keratosis. | 18q21.3 | |
| SERPINB8 | PI-8 | Intracellular | Possible inhibitor of furin. | 18q21.3 | |||
| SERPINB9 | PI-9 | Intracellular | Inhibitor of the cytotoxic granule protease granzyme B. | Knockout in mice causes immune dysfunction. | 6p25 | ||
| SERPINB10 | Bomapin | Intracellular | Unknown | Knockout in mice causes no obvious phenotype (C57/BL6; lab strain BC069938). | 18q21.3 | ||
| SERPINB11 | Intracellular | Unknown | Murine Serpinb11 is an active inhibitor whereas the human orthalogue is inactive. Deficiency in ponies is associated with hoof wall separation disease. | 18q21.3 | |||
| SERPINB12 | Yukopin | Intracellular | Unknown | 18q21.3 | |||
| SERPINB13 | Hurpin/Headpin | Intracellular | Inhibitor of papain-like cysteine proteases. | 18q21.3 | |||
| SERPINC1 | Antithrombin | Extracellular | Inhibitor of coagulation, specifically factor X, factor IX and thrombin. | Knockouts in mice are lethal. | Deficiency results in thrombosis and other clotting disorders (serpinopathy). | 1q23-q21 | 2ANT, 2ZNH, 1AZX, 1TB6, 2GD4, 1T1F |
| SERPIND1 | Heparin cofactor II | Extracellular | Inhibitor of thrombin. | Knockouts in mice are lethal. | 22q11 | 1JMJ, 1JMO | |
| SERPINE1 | Plasminogen activator inhibitor 1 | Extracellular | Inhibitor of thrombin, uPA and TPa. | 7q21.3-q22 | 1DVN, 1OC0 | ||
| SERPINE2 | Glia derived nexin / Protease nexin I | Extracellular | Inhibitor of uPA and tPA. | Abnormal expression leads to male infertility. Knockout in mice causes epilepsy. | 2q33-q35 | 4DY0 | |
| SERPINF1 | Pigment epithelium derived factor | Extracellular | Non-inhibitory, potent anti-angiogenic molecule. PEDF has been reported to bind the glycosaminoglycan hyaluronan. | Knockout in mice affects the vasculature and mass of the pancreas and the prostate. Promotes Notch–dependent renewal of adult periventricular neural stem cells. Mutations in humans cause osteogenesis imperfecta type VI. | 17p13.3 | 1IMV | |
| SERPINF2 | α2-antiplasmin | Extracellular | Inhibitor of plasmin, inhibitor of fibrinolysis. | Knockouts in mice show increased mice show increased fibrinolysis but no bleeding disorder. | Deficiency causes a rare bleeding disorder. | 17pter-p12 | 2R9Y |
| SERPING1 | Complement 1-inhibitor | Extracellular | Inhibitor of C1 esterase. | Several polymorphisms associated with macular degeneration and hereditary angeoedema. | 11q11-q13.1 | 2OAY | |
| SERPINH1 | 47 kDa Heat shock protein (HSP47) | Intracellular | Non-inhibitory, molecular chaperone in collagen folding. | Knockouts in mice are lethal. | Mutation in humans causes severe osteogenesis imperfecta. | 11p15 | 4AXY |
| SERPINI1 | Neuroserpin | Extracellular | Inhibitor of tPA, uPA and plasmin. | Mutation causes FENIB dementia (serpinopathy). | 3q26 | 1JJO, 3FGQ, 3F5N, 3F02 | |
| SERPINI2 | Pancpin | Extracellular | Unknown | Deficiency in mice causes pancreatic insufficiency via acinar cell loss. | 3q26 |
Specialised mammalian serpins
Many mammalian serpins have been identified that share no obvious orthology with a human serpin counterpart. Examples include numerous rodent serpins (particularly some of the murine intracellular serpins) as well as the uterine serpins. The term uterine serpin refers to members of the serpin A clade that are encoded by the SERPINA14 gene. Uterine serpins are produced by the endometrium of a restricted group of mammals in the Laurasiatheria clade under the influence of progesterone or estrogen. They are probably not functional proteinase inhibitors and may function during pregnancy to inhibit maternal immune responses against the conceptus or to participate in transplacental transport.
Insect
The Drosophila melanogaster genome contains 29 serpin encoding genes. Amino acid sequence analysis has placed 14 of these serpins in serpin clade Q and three in serpin clade K with the remaining twelve classified as orphan serpins not belonging to any clade. The clade classification system is difficult to use for Drosophila serpins and instead a nomenclature system has been adopted that is based on the position of serpin genes on the Drosophila chromosomes. Thirteen of the Drosophila serpins occur as isolated genes in the genome (including Serpin-27A, see below), with the remaining 16 organised into five gene clusters that occur at chromosome positions 28D (2 serpins), 42D (5 serpins), 43A (4 serpins), 77B (3 serpins) and 88E (2 serpins).
Studies on Drosophila serpins reveal that Serpin-27A inhibits the Easter protease (the final protease in the Nudel, Gastrulation Defective, Snake and Easter proteolytic cascade) and thus controls dorsoventral patterning. Easter functions to cleave Spätzle (a chemokine-type ligand), which results in toll-mediated signaling. As well as its central role in embryonic patterning, toll signaling is also important for the innate immune response in insects. Accordingly, serpin-27A also functions to control the insect immune response. In Tenebrio molitor (a large beetle), a protein (SPN93) comprising two discrete tandem serpin domains functions to regulate the toll proteolytic cascade.
Nematode
The genome of the nematode worm C. elegans contains 9 serpins, all of which lack signal sequences and so are likely intracellular. However, only 5 of these serpins appear to function as protease inhibitors. One, SRP-6, performs a protective function and guards against stress-induced calpain-associated lysosomal disruption. Further, SRP-6 inhibits lysosomal cysteine proteases released after lysosomal rupture. Accordingly, worms lacking SRP-6 are sensitive to stress. Most notably, SRP-6 knockout worms die when placed in water (the hypo-osmotic stress lethal phenotype or Osl). It has therefore been suggested that lysosomes play a general and controllable role in determining cell fate.
Plant
Plant serpins were amongst the first members of the superfamily that were identified. The serpin barley protein Z is highly abundant in barley grain, and one of the major protein components in beer. The genome of the model plant, Arabidopsis thaliana contain 18 serpin-like genes, although only 8 of these are full-length serpin sequences.
Plant serpins are potent inhibitors of mammalian chymotrypsin-like serine proteases in vitro, the best-studied example being barley serpin Zx (BSZx), which is able to inhibit trypsin and chymotrypsin as well as several blood coagulation factors. However, close relatives of chymotrypsin-like serine proteases are absent in plants. The RCL of several serpins from wheat grain and rye contain poly-Q repeat sequences similar to those present in the prolamin storage proteins of the endosperm. It has therefore been suggested that plant serpins may function to inhibit proteases from insects or microbes that would otherwise digest grain storage proteins. In support of this hypothesis, specific plant serpins have been identified in the phloem sap of pumpkin (CmPS-1) and cucumber plants. Although an inverse correlation between up-regulation of CmPS-1 expression and aphid survival was observed, in vitro feeding experiments revealed that recombinant CmPS-1 did not appear to affect insect survival.
Alternative roles and protease targets for plant serpins have been proposed. The Arabidopsis serpin, AtSerpin1 (At1g47710; 3LE2), mediates set-point control over programmed cell death by targeting the 'Responsive to Desiccation-21' (RD21) papain-like cysteine protease. AtSerpin1 also inhibits metacaspase-like proteases in vitro. Two other Arabidopsis serpins, AtSRP2 (At2g14540) and AtSRP3 (At1g64030) appear to be involved in responses to DNA damage.
Fungal
A single fungal serpin has been characterized to date: celpin from Piromyces spp. strain E2. Piromyces is a genus of anaerobic fungi found in the gut of ruminants and is important for digesting plant material. Celpin is predicted to be inhibitory and contains two N-terminal dockerin domains in addition to its serpin domain. Dockerins are commonly found in proteins that localise to the fungal cellulosome, a large extracellular multiprotein complex that breaks down cellulose. It is therefore suggested that celpin may protect the cellulosome against plant proteases. Certain bacterial serpins similarly localize to the cellulosome.
Prokaryotic
Predicted serpin genes are sporadically distributed in prokaryotes. In vitro studies on some of these molecules have revealed that they are able to inhibit proteases, and it is suggested that they function as inhibitors in vivo. Several prokaryote serpins are found in extremophiles. Accordingly, and in contrast to mammalian serpins, these molecules possess elevated resistance to heat denaturation. The precise role of most bacterial serpins remains obscure, although Clostridium thermocellum serpin localises to the cellulosome. It is suggested that the role of cellulosome-associated serpins may be to prevent unwanted protease activity against the cellulosome.
Viral
Serpins are also expressed by viruses as a way to evade the host's immune defense. In particular, serpins expressed by pox viruses, including cow pox (vaccinia) and rabbit pox (myxoma), are of interest because of their potential use as novel therapeutics for immune and inflammatory disorders as well as transplant therapy. Serp1 suppresses the TLR-mediated innate immune response and allows indefinite cardiac allograft survival in rats. Crma and Serp2 are both cross-class inhibitors and target both serine (granzyme B; albeit weakly) and cysteine proteases (caspase 1 and caspase 8). In comparison to their mammalian counterparts, viral serpins contain significant deletions of elements of secondary structure. Specifically, crmA lacks the D-helix as well as significant portions of the A- and E-helices.
External links
- PDB Molecule of the Month Serpin
- Merops protease inhibitor claudication (Family I4)
- Serpins at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- James Whisstock laboratory at Monash University
- Jim Huntington laboratory Archived 30 October 2016 at the Wayback Machine at University of Cambridge
- Frank Church laboratory at University of North Carolina at Chapel Hill
- Paul Declerck laboratory at Katholieke Universiteit Leuven
- Tom Roberts laboratory at University of Sydney
- Robert Fluhr laboratory at Weizmann Institute of Science
- Peter Gettins laboratory at University of Illinois at Chicago
- Overview of all the structural information available in the PDB for UniProt: P01009 (Human Alpha-1-antitrypsin) at the PDBe-KB.