Cystic fibrosis transmembrane conductance regulator (CFTR) is a membrane protein and anion channel in vertebrates that is encoded by the CFTR gene.
Geneticist Lap-Chee Tsui and his team identified the CFTR gene in 1989 as the gene linked with CF (Cystic Fibrosis).
The CFTR gene codes for an ABC transporter-class ion channel protein that conducts chloride and bicarbonate ions across epithelial cell membranes. Mutations of the CFTR gene affecting anion channel function lead to dysregulation of epithelial lining fluid (mucus) transport in the lung, pancreas and other organs, resulting in cystic fibrosis. Complications include thickened mucus in the lungs with frequent respiratory infections, and pancreatic insufficiency giving rise to malnutrition and diabetes. These conditions lead to chronic disability and reduced life expectancy. In male patients, the progressive obstruction and destruction of the developing vas deferens (spermatic cord) and epididymis appear to result from abnormal intraluminal secretions, causing congenital absence of the vas deferens and male infertility, and found associated with an imbalance of fatty acids.
Gene

The gene that encodes the human CFTR protein is found on chromosome 7, on the long arm at position q31.2. from base pair 116,907,253 to base pair 117,095,955. CFTR orthologs occur in the jawed vertebrates.
Each individual inherits two copies of the CFTR (cystic fibrosis transmembrane conductance regulator) gene. However, some of the inherited copies have been altered. So far, the CFTR gene has been associated with over 700 distinct mutations. An individual with CF inherits two defective copies of the CFTR gene. These mutations might be heterozygous, meaning they include two different mutations, and homozygous, meaning they involve the same mutation. Delta F508 is the most common mutation, accounting for more than 70% of all mutations. Those who are homozygous for Delta F508 are commonly affected by pancreatic insufficiency.
The CFTR gene has been used in animals as a nuclear DNA phylogenetic marker. Large genomic sequences of this gene have been used to explore the phylogeny of the major groups of mammals, and confirmed the grouping of placental orders into four major clades: Xenarthra, Afrotheria, Laurasiatheria, and Euarchonta plus Glires.
Mutations
Nearly 1000 cystic fibrosis-causing mutations have been described. The most common mutation, DeltaF508 (ΔF508) primarily known as a processing mutation which results from a deletion (Δ) of three nucleotides which results in a loss of the amino acid phenylalanine (F) at the 508th position on the protein. As a result, the protein does not fold normally and is more quickly degraded. The vast majority of mutations are infrequent. The distribution and frequency of mutations varies among different populations which has implications for genetic screening and counseling.
Drug discovery for therapeutics to address CF in all patients is complicated due to a large number of disease-causing mutations. Ideally, a library of cell lines and cell-based assays corresponding to all mutants is required to screen for broadly-active drug candidates. Cell engineering methods including fluorogenic oligonucleotide signaling probes may be used to detect and isolate clonal cell lines for each mutant.
Mutations consist of replacements, duplications, deletions or shortenings in the CFTR gene. This may result in proteins that may not function, work less effectively, are more quickly degraded, or are present in inadequate numbers.
It has been hypothesized that mutations in the CFTR gene may confer a selective advantage to heterozygous individuals. Cells expressing a mutant form of the CFTR protein are resistant to invasion by the Salmonella typhi bacterium, the agent of typhoid fever, and mice carrying a single copy of mutant CFTR are resistant to diarrhea caused by cholera toxin.
The most common mutations that cause cystic fibrosis and pancreatic insufficiency in humans are:
| Variant cDNA name (ordered 5' to 3') | Variant protein name | Variant legacy name | rsID | # alleles in CFTR2 | Allele frequency in CFTR2 | % pancreatic insufficient | Variant final determination (July 2020) |
| c.1521_1523delCTT | p.Phe508del | F508del | rs113993960 | 99061 | 0.69744 | 98% | CF-causing |
| c.1624G>T | p.Gly542X | G542X | rs113993959 | 3610 | 0.02542 | 98% | CF-causing |
| c.1652G>A | p.Gly551Asp | G551D | rs75527207 | 2986 | 0.02102 | 96% | CF-causing |
| c.3909C>G | p.Asn1303Lys | N1303K | rs80034486 | 2246 | 0.01581 | 98% | CF-causing |
| c.350G>A | p.Arg117His | R117H | rs78655421 | 1854 | 0.01305 | 23% | Varying clinical consequence |
| c.3846G>A | p.Trp1282X | W1282X | rs77010898 | 1726 | 0.01215 | 99% | CF-causing |
| c.489+1G>T | No protein name | 621+1G->T | rs78756941 | 1323 | 0.00931 | 99% | CF-causing |
| c.1657C>T | p.Arg553X | R553X | rs74597325 | 1323 | 0.00931 | 97% | CF-causing |
| c.1585-1G>A | No protein name | 1717-1G->A | rs76713772 | 1216 | 0.00856 | 97% | CF-causing |
| c.3718-2477C>T | No protein name | 3849+10kbC->T | rs75039782 | 1158 | 0.00815 | 33% | CF-causing |
| c.2657+5G>A | No protein name | 2789+5G->A | rs80224560 | 1027 | 0.00723 | 43% | CF-causing |
| c.1519_1521delATC | p. Ile507del | I507del | rs121908745 | 651 | 0.00458 | 98% | CF-causing |
| c.3484C>T | p.Arg1162X | R1162X | rs74767530 | 651 | 0.00458 | 97% | CF-causing |
| c.254G>A | p.Gly85Glu | G85E | rs75961395 | 616 | 0.00434 | 85% | CF-causing |
| c.3454G>C | p.Asp1152His | D1152H | rs75541969 | 571 | 0.00402 | 24% | Varying clinical consequence |
| c.2051_2052delAAinsG | p. Lys684SerfsX38 | 2183AA->G | rs121908799 | 542 | 0.00382 | 96% | CF-causing |
| c.3528delC | p. Lys1177SerfsX15 | 3659delC | rs121908747 | 539 | 0.00379 | 99% | CF-causing |
| c.1040G>C | p.Arg347Pro | R347P | rs77932196 | 533 | 0.00375 | 68% | CF-causing |
| c.1210−12T[5] | No protein name | 5T | rs1805177 | 516 | 0.00363 | 28% | Varying clinical consequence |
| c.2988+1G>A | No protein name | 3120+1G->A | rs75096551 | 501 | 0.00353 | 98% | CF-causing |
| c.1364C>A | p.Ala455Glu | A455E | rs74551128 | 500 | 0.00352 | 34% | CF-causing |
| c.3140-26A>G | No protein name | 3272-26A->G | rs76151804 | 470 | 0.00331 | 29% | CF-causing |
| c.1000C>T | p.Arg334Trp | R334W | rs121909011 | 429 | 0.00302 | 40% | CF-causing |
| c.1766+1G>A | No protein name | 1898+1G->A | rs121908748 | 421 | 0.00296 | 99% | CF-causing |
| c.54-5940_273+10250del21kb | p.Ser18ArgfsX16 | CFTRdele2,3 | not found | 417 | 0.00294 | 100% | CF-causing |
| c.1679G>C | p.Arg560Thr | R560T | rs80055610 | 343 | 0.00241 | 98% | CF-causing |
| c.617T>G | p. Leu206Trp | L206W | rs121908752 | 333 | 0.00234 | 20% | CF-causing |
| c.2052dupA | p.Gln685ThrfsX4 | 2184insA | rs121908786 | 329 | 0.00232 | 85% | CF-causing |
| c.262_263delTT | p. Leu88IlefsX22 | 394delTT | rs121908769 | 307 | 0.00216 | 97% | CF-causing |
| c.178G>T | p.Glu60X | E60X | rs77284892 | 296 | 0.00208 | 99% | CF-causing |
| c.1477C>T | p.Gln493X | Q493X | rs77101217 | 292 | 0.00206 | 98% | CF-causing |
| c.579+1G>T | No protein name | 711+1G->T | rs77188391 | 274 | 0.00193 | 98% | CF-causing |
| c.2052delA | p. Lys684AsnfsX38 | 2184delA | rs121908746 | 255 | 0.00180 | 98% | CF-causing |
| c.200C>T | p.Pro67Leu | P67L | rs368505753 | 239 | 0.00168 | 34% | CF-causing |
| c.3302T>A | p.Met1101Lys | M1101K | rs36210737 | 238 | 0.00168 | 69% | CF-causing |
| c.1408A>G | p.Met470Val | M470V | rs213950 | 235 | 0.00165 | 46% | Non CF-causing |
| c.3276C>A or c.3276C>G | p.Tyr1092X | Y1092X | rs121908761 | 225 | 0.00158 | 98% | CF-causing |
| c.3196C>T | p.Arg1066Cys | R1066C | rs78194216 | 220 | 0.00155 | 98% | CF-causing |
| c.1021_1022dupTC | p.Phe342HisfsX28 | 1154insTC | rs387906360 | 214 | 0.00151 | 99% | CF-causing |
| c.3773dupT | p. Leu1258PhefsX7 | 3905insT | rs121908789 | 210 | 0.00148 | 97% | CF-causing |
| c.1646G>A | p.Ser549Asn | S549N | rs121908755 | 203 | 0.00143 | 84% | CF-causing |
| c.1040G>A | p.Arg347His | R347H | rs77932196 | 199 | 0.00140 | 24% | CF-causing |
| c.948delT | p.Phe316LeufsX12 | 1078delT | rs121908744 | 184 | 0.00130 | 99% | CF-causing |
| c.1210-33_1210-6GT[12]T[4] | No protein name | 5T;TG12 | not found | 182 | 0.00128 | 14% | Varying clinical consequence |
| c.3472C>T | p.Arg1158X | R1158X | rs79850223 | 179 | 0.00126 | 99% | CF-causing |
| c.2834C>T | p.Ser945Leu | S945L | rs397508442 | 167 | 0.00118 | 40% | CF-causing |
| c.1558G>T | p. Val520Phe | V520F | rs77646904 | 156 | 0.00110 | 98% | CF-causing |
| c.443T>C | p. Ile148Thr | I148T | rs35516286 | 148 | 0.00104 | 88% | Non CF-causing |
| c.349C>T | p.Arg117Cys | R117C | rs77834169 | 146 | 0.00103 | 24% | CF-causing |
DeltaF508
DeltaF508 (ΔF508), full name CFTRΔF508 or F508del-CFTR (rs113993960), is a specific mutation within the CFTR gene involving deletion of three nucleotides spanning positions 507 and 508 of the CFTR gene on chromosome 7, which ultimately results in the loss of a single codon for the amino acid phenylalanine (F). A person with the CFTRΔF508 mutation will produce an abnormal CFTR protein that lacks this phenylalanine residue and which cannot fold properly. Most of this mutated protein does not escape the endoplasmic reticulum for further processing. The small amounts that reach the plasma membrane are destabilized and the anion channel opens infrequently. Having two copies of this mutation (one inherited from each parent) is by far the most common cause of cystic fibrosis (CF), responsible for nearly two-thirds of mutations worldwide.
Effects
The CFTR protein is largely expressed in cells of the pancreas, intestinal and respiratory epithelia, and all exocrine glands. When properly folded, it is shuttled to the cell membrane, where it becomes a transmembrane protein that forms aqueous channels allowing the flow of chloride and bicarbonate ions out of cells; it also simultaneously inhibits the uptake of sodium ions by another channel protein. Both of these functions help to maintain an ion gradient that causes osmosis to draw water out of the cells. The ΔF508 mutation leads to the misfolding of CFTR and its eventual degradation in the ER. In organisms with two complements of the mutation, the protein is almost entirely absent from the cell membrane, and these critical ion transport functions are not performed.
Having a homozygous pair of genes with the ΔF508 mutation prevents the CFTR protein from assuming its normal position in the cell membrane. This causes increased water retention in cells, corresponding dehydration of the extracellular space, and an associated cascade of effects on various parts of the body. These effects include: thicker mucous membranes in the epithelia of afflicted organs; obstruction of narrow respiratory airways as a result of thicker mucous and inhibition of the free movement of muco cilia; congenital absence of the vas deferens due to increased mucus thickness during fetal development; pancreatic insufficiency due to blockage of the pancreatic duct with mucus; and increased risk of respiratory infection due to build-up of thick, nutrient-rich mucus where bacteria thrive. These are the symptoms of cystic fibrosis, a genetic disorder; however, ΔF508 is not the only mutation that causes this disorder.
Being a heterozygous carrier (having a single copy of ΔF508) results in decreased water loss during diarrhea because malfunctioning or absent CFTR proteins cannot maintain stable ion gradients across cell membranes. Typicallnucleotide-binding-up of both Cl− and Na+ ions inside affected cells, creating a hypotonic solution outside the cells and causing water to diffuse into the cells by osmosis. Several studies indicate that heterozygous carriers are at increased risk for various symptoms. For example, it has been shown that heterozygosity for cystic fibrosis is associated with increased airway reactivity, and heterozygotes may be at risk for poor pulmonary function. Heterozygotes with wheeze have been shown to be at higher risk for poor pulmonary function or development and progression of chronic obstructive lung disease. One gene for cystic fibrosis is sufficient to produce mild lung abnormalities even in the absence of infection.
Mechanism
The CFTR gene is located on the long arm of chromosome 7, at position q31.2, and ultimately codes for a sequence of 1,480 amino acids. Normally, the three DNA base pairs A-T-C (paired with T-A-G on the opposite strand) at the gene's 507th position form the template for the mRNA codon A-U-C for isoleucine, while the three DNA base pairs T-T-T (paired with A-A-A) at the adjacent 508th position form the template for the codon U-U-U for phenylalanine. The ΔF508 mutation is a deletion of the C-G pair from position 507 along with the first two T-A pairs from position 508, leaving the DNA sequence A-T-T (paired with T-A-A) at position 507, which is transcribed into the mRNA codon A-U-U. Since A-U-U also codes for isoleucine, position 507's amino acid does not change, and the mutation's net effect is equivalent to a deletion ("Δ") of the sequence resulting in the codon for phenylalanine at position 508.
Prevalence
ΔF508 is present on at least one copy of chromosome 7 in approximately one in 30 Caucasians. Presence of the mutation on both copies causes the autosomal recessive disease cystic fibrosis. Scientists have estimated that the original mutation occurred over 52,000 years ago in Northern Europe though cystic fibrosis patients of other ethnicities are also known to harbor the mutation. The young allele age may be a consequence of past selection. One hypothesis as to why the otherwise detrimental mutation has been maintained by natural selection is that a single copy may present a positive effect by reducing water loss during cholera, though the introduction of pathogenic Vibrio cholerae into Europe did not occur until the late 18th century. Another theory posits that CF carriers (heterozygotes for ΔF508) are more resistant to typhoid fever, since CFTR has been shown to act as a receptor for Salmonella typhi bacteria to enter intestinal epithelial cells.
Cystic fibrosis ΔF508 heterozygotes may be overrepresented among individuals with asthma and may have poorer lung function than non-carriers. Carriers of a single CF mutation have a higher prevalence of chronic rhinosinusitis than the general population. Approximately 50% of cystic fibrosis cases in Europe are due to homozygous ΔF508 mutations (this varies widely by region), while the allele frequency of ΔF508 is about 70%. The remaining cases are caused by over 1,500 other mutations, including R117H, 1717-1G>A, and 2789+56G>A. These mutations, when combined with each other or even a single copy of ΔF508, may cause CF symptoms. The genotype is not strongly correlated with severity of the CF, though specific symptoms have been linked to certain mutations.
Structure

The CFTR gene is approximately 189 kb in length, with 27 exons and 26 introns. CFTR is a glycoprotein and is found on the surface of many epithelial cells in the body. CFTR consists of 5 domains, which include 2 transmembrane or membrane-spanning domains, 2 nucleotide-binding domains and a regulatory domain. The transmembrane domains are each connected to a nucleotide binding domain (NBD) in the cytoplasm. The first NBD is connected to the second transmembrane domain by a regulatory "R" domain that is a unique feature of CFTR, not present in other ABC transporters which carries 19 predicted sites for protein kinase A(PKA). Six of these have been reported to be phosphorylated in vivo. The ion channel only opens when its R-domain has been phosphorylated by PKA and ATP is bound at the NBDs. Phosphorylation displaces the disordered R domain from positions preventing NBD dimerization and opening. The amino-terminus is part of the lasso motif which anchors into the cell membrane. The carboxyl terminal of the protein is anchored to the cytoskeleton by a PDZ-interacting domain. The structure is shas(PDBitsI) shows a homopentameric assembly of mutated NBD1, the first nucleotide binding domain (NBD1) of the transporter
Location and function
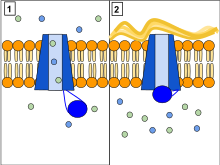
The CFTR gene is made up of 27 exons that encode its gene makeup and is found on the long (q) arm of chromosome 7 at locus 31.2. Exons are DNA fragments that provide the code for a protein structure. CFTR functions as phosphorylation and ATP-gated anion channel, increasing the conductance for certain anions (e.g. Cl−) to flow down their electrochemical gradient. ATP-driven conformational changes in CFTR open and close a gate to allow the transmembrane flow of anions down their electrochemical gradient. This in contrast to other ABC proteins, in which ATP-driven conformational changes fuel uphill substrate transport across cellular membranes. Essentially, CFTR is an ion channel that evolved as a 'broken' ABC transporter that leaks when in the open conformation.
CFTRs consist of five domains including two trans-membrane domains, each linked to a nucleotide-binding domain. CFTR also contains another domain called the regulatory domain. Other members of the ABC transporter superfamily are involved in the uptake of nutrients in prokaryotes, or in the export of a variety of substrates in eukaryotes. ABC transporters have evolved to transduce the free energy of ATP hydrolysis to the uphill movement of substrates across the cell membrane. They have two main conformations, one where the cargo binding site is facing the cytosol or inward facing (ATP free), and one where it is outward facing (ATP bound). ATP binds to each nucleotide-binding domain, which results in the subsequent NBD dimerization, leading to the rearrangement of the transmembrane helices. This changes the accessibility of the cargo binding site from an inward-facing position to an outward facing one. ATP binding, and the hydrolysis that follows, drives the alternative exposure of the cargo binding site, ensuring a unidirectional transport of cargo against an electrochemical gradient. In CFTR, alternating between an inward-facing conformation to an outward-facing one results in channel gating. In particular, NBD dimerization (favored by ATP binding) is coupled to transition to an outward-facing conformation in which an open transmembrane pathway for anions is formed. Subsequent hydrolysis (at the canonical active site, site 2, including Walker motifs of NBD2) destabilizes the NBD dimer and favors return to the inward-facing conformation, in which the anion permeation pathway is closed off.
The CFTR is found in the epithelial cells of many organs including the lung, liver, pancreas, digestive tract, and the female and male reproductive tracts.
In the airways of the lung, CFTR is most highly expressed by rare specialized cells called pulmonary ionocytes. In the skin, CFTR is strongly expressed in the sebaceous and eccrine sweat glands. In the eccrine glands, CFTR is located on the apical membrane of the epithelial cells that make up the duct of these sweat glands.
Normally, the protein allows movement of chloride, bicarbonate and thiocyanateions (with a negative charge) out of an epithelial cell into the Airway Surface Liquid and mucus. Positively charged sodium ions follow passively, increasing the total electrolyte concentration in the mucus, resulting in the movement of water out of the cell via osmosis.
In epithelial cells with motile cilia lining the bronchus and the oviduct, CFTR is located on the apical cell membrane but not on cilia. In contrast, ENaC (Epithelial sodium channel) is located along the entire length of the cilia.
In sweat glands, defective CFTR results in reduced transport of sodium chloride and sodium thiocyanate in the resorptive duct and therefore saltier sweat. This is the basis of a clinically important sweat test for cystic fibrosis often used diagnostically with genetic screening.
Interactions
Cystic fibrosis transmembrane conductance regulator has been shown to interact with:
It is inhibited by the anti-diarrhoea drug crofelemer.
Related conditions
- Congenital bilateral absence of vas deferens: Males with congenital bilateral absence of the vas deferens most often have a mild mutation (a change that allows partial function of the gene) in one copy of the CFTR gene and a cystic fibrosis-causing mutation in the other copy of CFTR.
- Cystic fibrosis: More than 1,800 mutations in the CFTR gene have been found but the majority of these have not been associated with cystic fibrosis. Most of these mutations either substitute one amino acid (a building block of proteins) for another amino acid in the CFTR protein or delete a small amount of DNA in the CFTR gene. The most common mutation, called ΔF508, is a deletion (Δ) of one amino acid (phenylalanine) at position 508 in the CFTR protein. This altered protein never reaches the cell membrane because it is degraded shortly after it is made. All disease-causing mutations in the CFTR gene prevent the channel from functioning properly, leading to a blockage of the movement of salt and water into and out of cells. As a result of this blockage, cells that line the passageways of the lungs, pancreas, and other organs produce abnormally thick, sticky mucus. This mucus obstructs the airways and glands, causing the characteristic signs and symptoms of cystic fibrosis. In addition, only thin mucus can be removed by cilia; thick mucus cannot, so it traps bacteria that give rise to chronic infections.
- Cholera: ADP-ribosylation caused by cholera toxin results in increased production of cyclic AMP which in turn opens the CFTR channel which leads to Over secretion of Cl−. Na+ and H2O follow Cl− into the small intestine, resulting in dehydration and loss of electrolytes.
Drug target
CFTR has been a drug target in efforts to find treatments for related conditions. Ivacaftor (trade name Kalydeco, developed as VX-770) is a drug approved by the FDA in 2012 for people with cystic fibrosis who have specific CFTR mutations. Ivacaftor was developed by Vertex Pharmaceuticals in conjunction with the Cystic Fibrosis Foundation and is the first drug that treats the underlying cause rather than the symptoms of the disease. Called "the most important new drug of 2012", and "a wonder drug" it is one of the most expensive drugs, costing over US$300,000 per year, which has led to criticism of Vertex for the high cost.
Further reading
- Kulczycki LL, Kostuch M, Bellanti JA (January 2003). "A clinical perspective of cystic fibrosis and new genetic findings: relationship of CFTR mutations to genotype-phenotype manifestations". American Journal of Medical Genetics. Part A. 116A (3): 262–267. doi:10.1002/ajmg.a.10886. PMID 12503104. S2CID 9245855.
- Vankeerberghen A, Cuppens H, Cassiman JJ (March 2002). "The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions". Journal of Cystic Fibrosis. 1 (1): 13–29. doi:10.1016/S1569-1993(01)00003-0. PMID 15463806.
- Tsui LC (1992). "Mutations and sequence variations detected in the cystic fibrosis transmembrane conductance regulator (CFTR) gene: a report from the Cystic Fibrosis Genetic Analysis Consortium". Human Mutation. 1 (3): 197–203. doi:10.1002/humu.1380010304. PMID 1284534. S2CID 35904538.
- McIntosh I, Cutting GR (July 1992). "Cystic fibrosis transmembrane conductance regulator and the etiology and pathogenesis of cystic fibrosis". FASEB Journal. 6 (10): 2775–2782. doi:10.1096/fasebj.6.10.1378801. PMID 1378801. S2CID 24932803.
- Drumm ML, Collins FS (1993). "Molecular biology of cystic fibrosis". Molecular Genetic Medicine. 3: 33–68. doi:10.1016/b978-0-12-462003-2.50006-7. ISBN 9780124620032. PMID 7693108.
- Kerem B, Kerem E (1996). "The molecular basis for disease variability in cystic fibrosis". European Journal of Human Genetics. 4 (2): 65–73. doi:10.1159/000472174. PMID 8744024. S2CID 41476164.
- Devidas S, Guggino WB (October 1997). "CFTR: domains, structure, and function". Journal of Bioenergetics and Biomembranes. 29 (5): 443–451. doi:10.1023/A:1022430906284. PMID 9511929. S2CID 6000695.
- Nagel G (December 1999). "Differential function of the two nucleotide binding domains on cystic fibrosis transmembrane conductance regulator". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1461 (2): 263–274. doi:10.1016/S0005-2736(99)00162-5. PMID 10581360.
- Boyle MP (2000). "Unique presentations and chronic complications in adult cystic fibrosis: do they teach us anything about CFTR?". Respiratory Research. 1 (3): 133–135. doi:10.1186/rr23. PMC 59552. PMID 11667976.
- Greger R, Schreiber R, Mall M, Wissner A, Hopf A, Briel M, et al. (2001). "Cystic fibrosis and CFTR". Pflügers Archiv. 443 Suppl 1: S3–S7. doi:10.1007/s004240100635. PMID 11845294. S2CID 8057614.
- Bradbury NA (2001). "cAMP signaling cascades and CFTR: is there more to learn?". Pflügers Archiv. 443 Suppl 1: S85–S91. doi:10.1007/s004240100651. PMID 11845310. S2CID 19373036.
- Dahan D, Evagelidis A, Hanrahan JW, Hinkson DA, Jia Y, Luo J, Zhu T (2001). "Regulation of the CFTR channel by phosphorylation". Pflügers Archiv. 443 Suppl 1: S92–S96. doi:10.1007/s004240100652. PMID 11845311. S2CID 8144727.
- Cohn JA, Noone PG, Jowell PS (September 2002). "Idiopathic pancreatitis related to CFTR: complex inheritance and identification of a modifier gene". Journal of Investigative Medicine. 50 (5): 247S–255S. doi:10.1136/jim-50-suppl5-01. PMID 12227654. S2CID 34017638.
- Schwartz M (February 2003). "[Cystic fibrosis transmembrane conductance regulator (CFTR) gene: mutations and clinical phenotypes]". Ugeskrift for Laeger. 165 (9): 912–916. PMID 12661515.
- Wong LJ, Alper OM, Wang BT, Lee MH, Lo SY (July 2003). "Two novel null mutations in a Taiwanese cystic fibrosis patient and a survey of East Asian CFTR mutations". American Journal of Medical Genetics. Part A. 120A (2): 296–298. doi:10.1002/ajmg.a.20039. PMID 12833420. S2CID 41060230.
- Cuppens H, Cassiman JJ (October 2004). "CFTR mutations and polymorphisms in male infertility". International Journal of Andrology. 27 (5): 251–256. doi:10.1111/j.1365-2605.2004.00485.x. PMID 15379964.
- Cohn JA, Mitchell RM, Jowell PS (March 2005). "The impact of cystic fibrosis and PSTI/SPINK1 gene mutations on susceptibility to chronic pancreatitis". Clinics in Laboratory Medicine. 25 (1): 79–100. doi:10.1016/j.cll.2004.12.007. PMID 15749233.
- Southern KW, Peckham D (2004). "Establishing a diagnosis of cystic fibrosis". Chronic Respiratory Disease. 1 (4): 205–210. doi:10.1191/1479972304cd044rs. PMID 16281647.
- Kandula L, Whitcomb DC, Lowe ME (June 2006). "Genetic issues in pediatric pancreatitis". Current Gastroenterology Reports. 8 (3): 248–253. doi:10.1007/s11894-006-0083-8. PMID 16764792. S2CID 23606613.
- Marcet B, Boeynaems JM (December 2006). "Relationships between cystic fibrosis transmembrane conductance regulator, extracellular nucleotides and cystic fibrosis". Pharmacology & Therapeutics. 112 (3): 719–732. doi:10.1016/j.pharmthera.2006.05.010. PMID 16828872.
- Wilschanski M, Durie PR (August 2007). "Patterns of GI disease in adulthood associated with mutations in the CFTR gene". Gut. 56 (8): 1153–1163. doi:10.1136/gut.2004.062786. PMC 1955522. PMID 17446304.
External links
- GeneReviews/NCBI/NIH/UW entry on CFTR-Related Disorders - Cystic Fibrosis (CF, Mucoviscidosis) and Congenital Absence of the Vas Deferens (CAVD)
- The Cystic Fibrosis Transmembrane Conductance Regulator Protein
- The Human Gene Mutation Database - CFTR Records
- Cystic Fibrosis Mutation Database
- Oak Ridge National Laboratory CFTR Information
- CFTR at OMIM (National Center for Biotechnology Information)
- Overview of all the structural information available in the PDB for UniProt: P13569 (Human Cystic fibrosis transmembrane conductance regulator) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: P26361 (Mouse Cystic fibrosis transmembrane conductance regulator) at the PDBe-KB.
|
PDB gallery
| |
|---|---|
|