
| Cascade (CRISPR-associated complex for antiviral defense) | |||||||
|---|---|---|---|---|---|---|---|
 CRISPR Cascade protein (cyan) bound to CRISPR RNA (green) and phage DNA (red)
| |||||||
| Identifiers | |||||||
| Organism | |||||||
| Symbol | CRISPR | ||||||
| Entrez | 947229 | ||||||
| PDB | 4QYZ | ||||||
| RefSeq (Prot) | NP_417241.1 | ||||||
| UniProt | P38036 | ||||||
| |||||||
| Part of a series on |
| CRISPR |
|---|
| Genome editing: CRISPR/Cas |
|
variants: Anti-CRISPR - CIRTS - CRISPeYCRISPR-Cas10 - CRISPR-Cas13 - CRISPR-BEST CRISPR-Display - CRISPR-Gold - CRISPRa - CRISPRi Easi-CRISPR - FACE |
| Enzyme |
|
Cas9 - FokI - EcoRI - PstI - SmaI HaeIII - Cas12a (Cpf1) - xCas9 |
| Applications |
| CAMERA - ICE - Genética dirigida |
| other Genome editing method: |
| Prime editing - Pro-AG - RESCUE - TALEN - ZFN - LEAPER |
| Part of a series on |
| Genetic engineering |
|---|
| Genetically modified organisms |
| History and regulation |
| Process |
| Applications |
| Controversies |
CRISPR (/ˈkrɪspər/) (an acronym for clustered regularly interspaced short palindromic repeats) is a family of DNA sequences found in the genomes of prokaryotic organisms such as bacteria and archaea. These sequences are derived from DNA fragments of bacteriophages that had previously infected the prokaryote. They are used to detect and destroy DNA from similar bacteriophages during subsequent infections. Hence these sequences play a key role in the antiviral (i.e. anti-phage) defense system of prokaryotes and provide a form of acquired immunity. CRISPR is found in approximately 50% of sequenced bacterial genomes and nearly 90% of sequenced archaea.

Cas9 (or "CRISPR-associated protein 9") is an enzyme that uses CRISPR sequences as a guide to recognize and cleave specific strands of DNA that are complementary to the CRISPR sequence. Cas9 enzymes together with CRISPR sequences form the basis of a technology known as CRISPR-Cas9 that can be used to edit genes within organisms. This editing process has a wide variety of applications including basic biological research, development of biotechnological products, and treatment of diseases. The development of the CRISPR-Cas9 genome editing technique was recognized by the Nobel Prize in Chemistry in 2020 which was awarded to Emmanuelle Charpentier and Jennifer Doudna.
In 2022, in a proceeding at the United States Patent and Trademark Office (interference 106,115), the Patent Trial and Appeal Board decided that the inventor for the US patent covering application of CRISPR-Cas9 in eukaryotic cells is Feng Zhang, a professor of the Broad Institute.
History
Repeated sequences
The discovery of clustered DNA repeats took place independently in three parts of the world. The first description of what would later be called CRISPR is from Osaka University researcher Yoshizumi Ishino and his colleagues in 1987. They accidentally cloned part of a CRISPR sequence together with the "iap" gene (isozyme conversion of alkaline phosphatase) from the genome of Escherichia coli which was their target. The organization of the repeats was unusual. Repeated sequences are typically arranged consecutively, without interspersing different sequences. They did not know the function of the interrupted clustered repeats.
In 1993, researchers of Mycobacterium tuberculosis in the Netherlands published two articles about a cluster of interrupted direct repeats (DR) in that bacterium. They recognized the diversity of the sequences that intervened in the direct repeats among different strains of M. tuberculosis and used this property to design a typing method that was named spoligotyping, which is still in use, today.
Francisco Mojica at the University of Alicante in Spain studied repeats observed in the archaeal organisms of Haloferax and Haloarcula species and their function. Mojica's supervisor surmised at the time that the clustered repeats had a role in correctly segregating replicated DNA into daughter cells during cell division because plasmids and chromosomes with identical repeat arrays could not coexist in Haloferax volcanii. Transcription of the interrupted repeats was also noted for the first time; this was the first full characterization of CRISPR. By 2000, Mojica performed a survey of scientific literature and one of his students performed a search in published genomes with a program devised by himself. They identified interrupted repeats in 20 species of microbes as belonging to the same family. Because those sequences were interspaced, Mojica initially called these sequences "short regularly spaced repeats" (SRSR). In 2001, Mojica and Ruud Jansen, who were searching for additional interrupted repeats, proposed the acronym CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) to alleviate the confusion stemming from the numerous acronyms used to describe the sequences in the scientific literature. In 2002, Tang, et al. showed evidence that CRISPR repeat regions from the genome of Archaeoglobus fulgidus were transcribed into long RNA molecules that were subsequently processed into unit-length small RNAs, plus some longer forms of 2, 3, or more spacer-repeat units.
In 2005, yogurt researcher Rodolphe Barrangou discovered that Streptococcus thermophilus, after iterative phage challenges, develops increased phage resistance, and this enhanced resistance is due to the incorporation of additional CRISPR spacer sequences. The Danish food company Danisco, which at that time Barrangou worked for, then developed phage-resistant S. thermophilus strains for use in yogurt production. Danisco was later bought out by DuPont, which "owns about 50 percent of the global dairy culture market" and the technology went mainstream.
CRISPR-associated systems
A major addition to the understanding of CRISPR came with Jansen's observation that the prokaryote repeat cluster was accompanied by a set of homologous genes that make up CRISPR-associated systems or cas genes. Four cas genes (cas 1–4) were initially recognized. The Cas proteins showed helicase and nuclease motifs, suggesting a role in the dynamic structure of the CRISPR loci. In this publication, the acronym CRISPR was used as the universal name of this pattern. However, the CRISPR function remained enigmatic.

In 2005, three independent research groups showed that some CRISPR spacers are derived from phage DNA and extrachromosomal DNA such as plasmids. In effect, the spacers are fragments of DNA gathered from viruses that previously tried to attack the cell. The source of the spacers was a sign that the CRISPR/cas system could have a role in adaptive immunity in bacteria. All three studies proposing this idea were initially rejected by high-profile journals, but eventually appeared in other journals.
The first publication proposing a role of CRISPR-Cas in microbial immunity, by Mojica and collaborators at the University of Alicante, predicted a role for the RNA transcript of spacers on target recognition in a mechanism that could be analogous to the RNA interference system used by eukaryotic cells. Koonin and colleagues extended this RNA interference hypothesis by proposing mechanisms of action for the different CRISPR-Cas subtypes according to the predicted function of their proteins.
Experimental work by several groups revealed the basic mechanisms of CRISPR-Cas immunity. In 2007, the first experimental evidence that CRISPR was an adaptive immune system was published. A CRISPR region in Streptococcus thermophilus acquired spacers from the DNA of an infecting bacteriophage. The researchers manipulated the resistance of S. thermophilus to different types of phages by adding and deleting spacers whose sequence matched those found in the tested phages. In 2008, Brouns and Van der Oost identified a complex of Cas proteins (called Cascade) that in E. coli cut the CRISPR RNA precursor within the repeats into mature spacer-containing RNA molecules called CRISPR RNA (crRNA), which remained bound to the protein complex. Moreover, it was found that Cascade, crRNA and a helicase/nuclease (Cas3) were required to provide a bacterial host with immunity against infection by a DNA virus. By designing an anti-virus CRISPR, they demonstrated that two orientations of the crRNA (sense/antisense) provided immunity, indicating that the crRNA guides were targeting dsDNA. That year Marraffini and Sontheimer confirmed that a CRISPR sequence of S. epidermidis targeted DNA and not RNA to prevent conjugation. This finding was at odds with the proposed RNA-interference-like mechanism of CRISPR-Cas immunity, although a CRISPR-Cas system that targets foreign RNA was later found in Pyrococcus furiosus. A 2010 study showed that CRISPR-Cas cuts both strands of phage and plasmid DNA in S. thermophilus.
Cas9
A simpler CRISPR system from Streptococcus pyogenes relies on the protein Cas9. The Cas9 endonuclease is a four-component system that includes two small molecules: crRNA and trans-activating CRISPR RNA (tracrRNA).Jennifer Doudna and Emmanuelle Charpentier re-engineered the Cas9 endonuclease into a more manageable two-component system by fusing the two RNA molecules into a "single-guide RNA" that, when combined with Cas9, could find and cut the DNA target specified by the guide RNA. This contribution was so significant that it was recognized by the Nobel Prize in Chemistry in 2020. By manipulating the nucleotide sequence of the guide RNA, the artificial Cas9 system could be programmed to target any DNA sequence for cleavage. Another group of collaborators comprising Virginijus Šikšnys together with Gasiūnas, Barrangou, and Horvath showed that Cas9 from the S. thermophilus CRISPR system can also be reprogrammed to target a site of their choosing by changing the sequence of its crRNA. These advances fueled efforts to edit genomes with the modified CRISPR-Cas9 system.
Groups led by Feng Zhang and George Church simultaneously published descriptions of genome editing in human cell cultures using CRISPR-Cas9 for the first time. It has since been used in a wide range of organisms, including baker's yeast (Saccharomyces cerevisiae), the opportunistic pathogen Candida albicans, zebrafish (Danio rerio), fruit flies (Drosophila melanogaster), ants (Harpegnathos saltator and Ooceraea biroi), mosquitoes (Aedes aegypti), nematodes (Caenorhabditis elegans), plants, mice (Mus musculus domesticus), monkeys and human embryos.
CRISPR has been modified to make programmable transcription factors that allows targeting and activation or silencing specific genes.

The CRISPR-Cas9 system has shown to make effective gene edits in Human tripronuclear zygotes first described in a 2015 paper by Chinese scientists P. Liang and Y. Xu. The system made a successful cleavage of mutant Beta-Hemoglobin (HBB) in 28 out of 54 embryos. 4 out of the 28 embryos were successfully recombined using a donor template given by the scientists. The scientists showed that during DNA recombination of the cleaved strand, the homologous endogenous sequence HBD competes with the exogenous donor template. DNA repair in human embryos is much more complicated and particular than in derived stem cells.
Cas12a
In 2015, the nuclease Cas12a (formerly known as Cpf1) was characterized in the CRISPR/Cpf1 system of the bacterium Francisella novicida. Its original name, from a TIGRFAMs protein family definition built in 2012, reflects the prevalence of its CRISPR-Cas subtype in the Prevotella and Francisella lineages. Cas12a showed several key differences from Cas9 including: causing a 'staggered' cut in double stranded DNA as opposed to the 'blunt' cut produced by Cas9, relying on a 'T rich' PAM (providing alternative targeting sites to Cas9), and requiring only a CRISPR RNA (crRNA) for successful targeting. By contrast, Cas9 requires both crRNA and a transactivating crRNA (tracrRNA).
These differences may give Cas12a some advantages over Cas9. For example, Cas12a's small crRNAs are ideal for multiplexed genome editing, as more of them can be packaged in one vector than can Cas9's sgRNAs. The sticky 5′ overhangs left by Cas12a can also be used for DNA assembly that is much more target-specific than traditional restriction enzyme cloning. Finally, Cas12a cleaves DNA 18–23 base pairs downstream from the PAM site. This means there is no disruption to the recognition sequence after repair, and so Cas12a enables multiple rounds of DNA cleavage. By contrast, since Cas9 cuts only 3 base pairs upstream of the PAM site, the NHEJ pathway results in indel mutations that destroy the recognition sequence, thereby preventing further rounds of cutting. In theory, repeated rounds of DNA cleavage should cause an increased opportunity for the desired genomic editing to occur. A distinctive feature of Cas12a, as compared to Cas9, is that after cutting its target, Cas12a remains bound to the target and then cleaves other ssDNA molecules non-discriminately. This property is called "collateral cleavage" or "trans-cleavage" activity and has been exploited for the development of various diagnostic technologies.
Cas13
In 2016, the nuclease Cas13a (formerly known as C2c2) from the bacterium Leptotrichia shahii was characterized. Cas13 is an RNA-guided RNA endonuclease, which means that it does not cleave DNA, but only single-stranded RNA. Cas13 is guided by its crRNA to a ssRNA target and binds and cleaves the target. Similar to Cas12a, the Cas13 remains bound to the target and then cleaves other ssRNA molecules non-discriminately. This collateral cleavage property has been exploited for the development of various diagnostic technologies.
In 2021, Dr. Hui Yang characterized novel miniature Cas13 protein (mCas13) variants, Cas13X & Cas13Y. Using a small portion of N gene sequence from SARS-CoV-2 as a target in characterization of mCas13, revealed the sensitivity and specificity of mCas13 coupled with RT-LAMP for detection of SARS-CoV-2 in both synthetic and clinical samples over other available standard tests like RT-qPCR (1 copy/μL).
Locus structure
Repeats and spacers
The CRISPR array is made up of an AT-rich leader sequence followed by short repeats that are separated by unique spacers. CRISPR repeats typically range in size from 28 to 37 base pairs (bps), though there can be as few as 23 bp and as many as 55 bp. Some show dyad symmetry, implying the formation of a secondary structure such as a stem-loop ('hairpin') in the RNA, while others are designed to be unstructured. The size of spacers in different CRISPR arrays is typically 32 to 38 bp (range 21 to 72 bp). New spacers can appear rapidly as part of the immune response to phage infection. There are usually fewer than 50 units of the repeat-spacer sequence in a CRISPR array.
CRISPR RNA structures








Cas genes and CRISPR subtypes
Small clusters of cas genes are often located next to CRISPR repeat-spacer arrays. Collectively the 93 cas genes are grouped into 35 families based on sequence similarity of the encoded proteins. 11 of the 35 families form the cas core, which includes the protein families Cas1 through Cas9. A complete CRISPR-Cas locus has at least one gene belonging to the cas core.
CRISPR-Cas systems fall into two classes. Class 1 systems use a complex of multiple Cas proteins to degrade foreign nucleic acids. Class 2 systems use a single large Cas protein for the same purpose. Class 1 is divided into types I, III, and IV; class 2 is divided into types II, V, and VI. The 6 system types are divided into 19 subtypes. Each type and most subtypes are characterized by a "signature gene" found almost exclusively in the category. Classification is also based on the complement of cas genes that are present. Most CRISPR-Cas systems have a Cas1 protein. The phylogeny of Cas1 proteins generally agrees with the classification system, but exceptions exist due to module shuffling. Many organisms contain multiple CRISPR-Cas systems suggesting that they are compatible and may share components. The sporadic distribution of the CRISPR/Cas subtypes suggests that the CRISPR/Cas system is subject to horizontal gene transfer during microbial evolution.
| Class | Cas type | Cas subtype | Signature protein | Function | Reference |
|---|---|---|---|---|---|
| 1 | I | — | Cas3 | Single-stranded DNA nuclease (HD domain) and ATP-dependent helicase | |
| I-A | Cas8a, Cas5 | Cas8 is a Subunit of the interference module that is important in targeting of invading DNA by recognizing the PAM sequence. Cas5 is required for processing and stability of crRNAs | |||
| I-B | Cas8b | ||||
| I-C | Cas8c | ||||
| I-D | Cas10d | contains a domain homologous to the palm domain of nucleic acid polymerases and nucleotide cyclases | |||
| I-E | Cse1, Cse2 | ||||
| I-F | Csy1, Csy2, Csy3 | Not determined | |||
| I-G | GSU0054 | ||||
| III | — | Cas10 | Homolog of Cas10d and Cse1. Binds CRISPR target RNA and promotes stability of the interference complex | ||
| III-A | Csm2 | Not Determined | |||
| III-B | Cmr5 | Not Determined | |||
| III-C | Cas10 or Csx11 | ||||
| III-D | Csx10 | ||||
| III-E | |||||
| III-F | |||||
| IV | — | Csf1 | |||
| IV-A | |||||
| IV-B | |||||
| IV-C | |||||
| 2 | II | — | Cas9 | Nucleases RuvC and HNH together produce DSBs, and separately can produce single-strand breaks. Ensures the acquisition of functional spacers during adaptation. | |
| II-A | Csn2 | Ring-shaped DNA-binding protein. Involved in primed adaptation in Type II CRISPR system. | |||
| II-B | Cas4 | Endonuclease that works with cas1 and cas2 to generate spacer sequences | |||
| II-C | Characterized by the absence of either Csn2 or Cas4 | ||||
| V | — | Cas12 | Nuclease RuvC. Lacks HNH. | ||
| V-A | Cas12a (Cpf1) | Auto-processing pre-crRNA activity for multiplex gene regulation | |||
| V-B | Cas12b (C2c1) | ||||
| V-C | Cas12c (C2c3) | ||||
| V-D | Cas12d (CasY) | ||||
| V-E | Cas12e (CasX) | ||||
| V-F | Cas12f (Cas14, C2c10) | ||||
| V-G | Cas12g | ||||
| V-H | Cas12h | ||||
| V-I | Cas12i | ||||
| V-K | Cas12k (C2c5) | ||||
| V-U | C2c4, C2c8, C2c9 | ||||
| VI | — | Cas13 | RNA-guided RNase | ||
| VI-A | Cas13a (C2c2) | ||||
| VI-B | Cas13b | ||||
| VI-C | Cas13c | ||||
| VI-D | Cas13d | ||||
| VI-X | Cas13x.1 | RNA dependent RNA polymerase, Prophylactic RNA-virus inhibition | |||
| VI-Y |
Mechanism

(1) Acquisition begins by recognition of invading DNA by Cas1 and Cas2 and cleavage of a protospacer.
(2) The protospacer is ligated to the direct repeat adjacent to the leader sequence and
(3) single strand extension repairs the CRISPR and duplicates the direct repeat. The crRNA processing and interference stages occur differently in each of the three major CRISPR systems.
(4) The primary CRISPR transcript is cleaved by cas genes to produce crRNAs.
(5) In type I systems Cas6e/Cas6f cleave at the junction of ssRNA and dsRNA formed by hairpin loops in the direct repeat. Type II systems use a trans-activating (tracr) RNA to form dsRNA, which is cleaved by Cas9 and RNaseIII. Type III systems use a Cas6 homolog that does not require hairpin loops in the direct repeat for cleavage.
(6) In type II and type III systems secondary trimming is performed at either the 5’ or 3’ end to produce mature crRNAs.
(7) Mature crRNAs associate with Cas proteins to form interference complexes.
(8) In type I and type II systems, interactions between the protein and PAM sequence are required for degradation of invading DNA. Type III systems do not require a PAM for successful degradation and in type III-A systems basepairing occurs between the crRNA and mRNA rather than the DNA, targeted by type III-B systems.




CRISPR-Cas immunity is a natural process of bacteria and archaea. CRISPR-Cas prevents bacteriophage infection, conjugation and natural transformation by degrading foreign nucleic acids that enter the cell.
Spacer acquisition
When a microbe is invaded by a bacteriophage, the first stage of the immune response is to capture phage DNA and insert it into a CRISPR locus in the form of a spacer. Cas1 and Cas2 are found in both types of CRISPR-Cas immune systems, which indicates that they are involved in spacer acquisition. Mutation studies confirmed this hypothesis, showing that removal of cas1 or cas2 stopped spacer acquisition, without affecting CRISPR immune response.
Multiple Cas1 proteins have been characterised and their structures resolved. Cas1 proteins have diverse amino acid sequences. However, their crystal structures are similar and all purified Cas1 proteins are metal-dependent nucleases/integrases that bind to DNA in a sequence-independent manner. Representative Cas2 proteins have been characterised and possess either (single strand) ssRNA- or (double strand) dsDNA- specific endoribonuclease activity.
In the I-E system of E. coli Cas1 and Cas2 form a complex where a Cas2 dimer bridges two Cas1 dimers. In this complex Cas2 performs a non-enzymatic scaffolding role, binding double-stranded fragments of invading DNA, while Cas1 binds the single-stranded flanks of the DNA and catalyses their integration into CRISPR arrays. New spacers are usually added at the beginning of the CRISPR next to the leader sequence creating a chronological record of viral infections. In E. coli a histone like protein called integration host factor (IHF), which binds to the leader sequence, is responsible for the accuracy of this integration. IHF also enhances integration efficiency in the type I-F system of Pectobacterium atrosepticum. but in other systems, different host factors may be required
Protospacer adjacent motifs (PAM)
Bioinformatic analysis of regions of phage genomes that were excised as spacers (termed protospacers) revealed that they were not randomly selected but instead were found adjacent to short (3–5 bp) DNA sequences termed protospacer adjacent motifs (PAM). Analysis of CRISPR-Cas systems showed PAMs to be important for type I and type II, but not type III systems during acquisition. In type I and type II systems, protospacers are excised at positions adjacent to a PAM sequence, with the other end of the spacer cut using a ruler mechanism, thus maintaining the regularity of the spacer size in the CRISPR array. The conservation of the PAM sequence differs between CRISPR-Cas systems and appears to be evolutionarily linked to Cas1 and the leader sequence.
New spacers are added to a CRISPR array in a directional manner, occurring preferentially, but not exclusively, adjacent to the leader sequence. Analysis of the type I-E system from E. coli demonstrated that the first direct repeat adjacent to the leader sequence is copied, with the newly acquired spacer inserted between the first and second direct repeats.
The PAM sequence appears to be important during spacer insertion in type I-E systems. That sequence contains a strongly conserved final nucleotide (nt) adjacent to the first nt of the protospacer. This nt becomes the final base in the first direct repeat. This suggests that the spacer acquisition machinery generates single-stranded overhangs in the second-to-last position of the direct repeat and in the PAM during spacer insertion. However, not all CRISPR-Cas systems appear to share this mechanism as PAMs in other organisms do not show the same level of conservation in the final position. It is likely that in those systems, a blunt end is generated at the very end of the direct repeat and the protospacer during acquisition.
Insertion variants
Analysis of Sulfolobus solfataricus CRISPRs revealed further complexities to the canonical model of spacer insertion, as one of its six CRISPR loci inserted new spacers randomly throughout its CRISPR array, as opposed to inserting closest to the leader sequence.
Multiple CRISPRs contain many spacers to the same phage. The mechanism that causes this phenomenon was discovered in the type I-E system of E. coli. A significant enhancement in spacer acquisition was detected where spacers already target the phage, even mismatches to the protospacer. This ‘priming’ requires the Cas proteins involved in both acquisition and interference to interact with each other. Newly acquired spacers that result from the priming mechanism are always found on the same strand as the priming spacer. This observation led to the hypothesis that the acquisition machinery slides along the foreign DNA after priming to find a new protospacer.
Biogenesis
CRISPR-RNA (crRNA), which later guides the Cas nuclease to the target during the interference step, must be generated from the CRISPR sequence. The crRNA is initially transcribed as part of a single long transcript encompassing much of the CRISPR array. This transcript is then cleaved by Cas proteins to form crRNAs. The mechanism to produce crRNAs differs among CRISPR/Cas systems. In type I-E and type I-F systems, the proteins Cas6e and Cas6f respectively, recognise stem-loops created by the pairing of identical repeats that flank the crRNA. These Cas proteins cleave the longer transcript at the edge of the paired region, leaving a single crRNA along with a small remnant of the paired repeat region.
Type III systems also use Cas6, however, their repeats do not produce stem-loops. Cleavage instead occurs by the longer transcript wrapping around the Cas6 to allow cleavage just upstream of the repeat sequence.
Type II systems lack the Cas6 gene and instead utilize RNaseIII for cleavage. Functional type II systems encode an extra small RNA that is complementary to the repeat sequence, known as a trans-activating crRNA (tracrRNA). Transcription of the tracrRNA and the primary CRISPR transcript results in base pairing and the formation of dsRNA at the repeat sequence, which is subsequently targeted by RNaseIII to produce crRNAs. Unlike the other two systems, the crRNA does not contain the full spacer, which is instead truncated at one end.
CrRNAs associate with Cas proteins to form ribonucleotide complexes that recognize foreign nucleic acids. CrRNAs show no preference between the coding and non-coding strands, which is indicative of an RNA-guided DNA-targeting system. The type I-E complex (commonly referred to as Cascade) requires five Cas proteins bound to a single crRNA.
Interference
During the interference stage in type I systems, the PAM sequence is recognized on the crRNA-complementary strand and is required along with crRNA annealing. In type I systems correct base pairing between the crRNA and the protospacer signals a conformational change in Cascade that recruits Cas3 for DNA degradation.
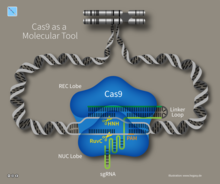
Type II systems rely on a single multifunctional protein, Cas9, for the interference step.Cas9 requires both the crRNA and the tracrRNA to function and cleave DNA using its dual HNH and RuvC/RNaseH-like endonuclease domains. Basepairing between the PAM and the phage genome is required in type II systems. However, the PAM is recognized on the same strand as the crRNA (the opposite strand to type I systems).
Type III systems, like type I require six or seven Cas proteins binding to crRNAs. The type III systems analysed from S. solfataricus and P. furiosus both target the mRNA of phages rather than phage DNA genome, which may make these systems uniquely capable of targeting RNA-based phage genomes. Type III systems were also found to target DNA in addition to RNA using a different Cas protein in the complex, Cas10. The DNA cleavage was shown to be transcription dependent.
The mechanism for distinguishing self from foreign DNA during interference is built into the crRNAs and is therefore likely common to all three systems. Throughout the distinctive maturation process of each major type, all crRNAs contain a spacer sequence and some portion of the repeat at one or both ends. It is the partial repeat sequence that prevents the CRISPR-Cas system from targeting the chromosome as base pairing beyond the spacer sequence signals self and prevents DNA cleavage. RNA-guided CRISPR enzymes are classified as type V restriction enzymes.
Evolution
| CRISPR associated protein Cas2 (adaptation RNase) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 crystal structure of a hypothetical protein tt1823 from Thermus thermophilus
| |||||||||
| Identifiers | |||||||||
| Symbol | CRISPR_Cas2 | ||||||||
| Pfam | PF09827 | ||||||||
| InterPro | IPR019199 | ||||||||
| CDD | cd09638 | ||||||||
| |||||||||
| CRISPR-associated protein CasA/Cse1 (Type I effector DNase) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | CRISPR_Cse1 | ||||||||
| Pfam | PF09481 | ||||||||
| InterPro | IPR013381 | ||||||||
| CDD | cd09729 | ||||||||
| |||||||||
| CRISPR associated protein CasC/Cse3/Cas6 (Type I effector RNase) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 crystal structure of a crispr-associated protein from Thermus thermophilus
| |||||||||
| Identifiers | |||||||||
| Symbol | CRISPR_assoc | ||||||||
| Pfam | PF08798 | ||||||||
| Pfam clan | CL0362 | ||||||||
| InterPro | IPR010179 | ||||||||
| CDD | cd09727 | ||||||||
| |||||||||
The cas genes in the adaptor and effector modules of the CRISPR-Cas system are believed to have evolved from two different ancestral modules. A transposon-like element called casposon encoding the Cas1-like integrase and potentially other components of the adaptation module was inserted next to the ancestral effector module, which likely functioned as an independent innate immune system. The highly conserved cas1 and cas2 genes of the adaptor module evolved from the ancestral module while a variety of class 1 effector cas genes evolved from the ancestral effector module. The evolution of these various class 1 effector module cas genes was guided by various mechanisms, such as duplication events. On the other hand, each type of class 2 effector module arose from subsequent independent insertions of mobile genetic elements. These mobile genetic elements took the place of the multiple gene effector modules to create single gene effector modules that produce large proteins which perform all the necessary tasks of the effector module. The spacer regions of CRISPR-Cas systems are taken directly from foreign mobile genetic elements and thus their long term evolution is hard to trace. The non-random evolution of these spacer regions has been found to be highly dependent on the environment and the particular foreign mobile genetic elements it contains.
CRISPR/Cas can immunize bacteria against certain phages and thus halt transmission. For this reason, Koonin described CRISPR/Cas as a Lamarckian inheritance mechanism. However, this was disputed by a critic who noted, "We should remember [Lamarck] for the good he contributed to science, not for things that resemble his theory only superficially. Indeed, thinking of CRISPR and other phenomena as Lamarckian only obscures the simple and elegant way evolution really works". But as more recent studies have been conducted, it has become apparent that the acquired spacer regions of CRISPR-Cas systems are indeed a form of Lamarckian evolution because they are genetic mutations that are acquired and then passed on. On the other hand, the evolution of the Cas gene machinery that facilitates the system evolves through classic Darwinian evolution.
Coevolution
Analysis of CRISPR sequences revealed coevolution of host and viral genomes. Cas9 proteins are highly enriched in pathogenic and commensal bacteria. CRISPR/Cas-mediated gene regulation may contribute to the regulation of endogenous bacterial genes, particularly during interaction with eukaryotic hosts. For example, Francisella novicida uses a unique, small, CRISPR/Cas-associated RNA (scaRNA) to repress an endogenous transcript encoding a bacterial lipoprotein that is critical for F. novicida to dampen host response and promote virulence.
The basic model of CRISPR evolution is newly incorporated spacers driving phages to mutate their genomes to avoid the bacterial immune response, creating diversity in both the phage and host populations. To resist a phage infection, the sequence of the CRISPR spacer must correspond perfectly to the sequence of the target phage gene. Phages can continue to infect their hosts' given point mutations in the spacer. Similar stringency is required in PAM or the bacterial strain remains phage sensitive.
Rates
A study of 124 S. thermophilus strains showed that 26% of all spacers were unique and that different CRISPR loci showed different rates of spacer acquisition. Some CRISPR loci evolve more rapidly than others, which allowed the strains' phylogenetic relationships to be determined. A comparative genomic analysis showed that E. coli and S. enterica evolve much more slowly than S. thermophilus. The latter's strains that diverged 250 thousand years ago still contained the same spacer complement.
Metagenomic analysis of two acid-mine-drainage biofilms showed that one of the analyzed CRISPRs contained extensive deletions and spacer additions versus the other biofilm, suggesting a higher phage activity/prevalence in one community than the other. In the oral cavity, a temporal study determined that 7–22% of spacers were shared over 17 months within an individual while less than 2% were shared across individuals.
From the same environment, a single strain was tracked using PCR primers specific to its CRISPR system. Broad-level results of spacer presence/absence showed significant diversity. However, this CRISPR added 3 spacers over 17 months, suggesting that even in an environment with significant CRISPR diversity some loci evolve slowly.
CRISPRs were analysed from the metagenomes produced for the human microbiome project. Although most were body-site specific, some within a body site are widely shared among individuals. One of these loci originated from streptococcal species and contained ≈15,000 spacers, 50% of which were unique. Similar to the targeted studies of the oral cavity, some showed little evolution over time.
CRISPR evolution was studied in chemostats using S. thermophilus to directly examine spacer acquisition rates. In one week, S. thermophilus strains acquired up to three spacers when challenged with a single phage. During the same interval, the phage developed single nucleotide polymorphisms that became fixed in the population, suggesting that targeting had prevented phage replication absent these mutations.
Another S. thermophilus experiment showed that phages can infect and replicate in hosts that have only one targeting spacer. Yet another showed that sensitive hosts can exist in environments with high phage titres. The chemostat and observational studies suggest many nuances to CRISPR and phage (co)evolution.
Identification
CRISPRs are widely distributed among bacteria and archaea and show some sequence similarities. Their most notable characteristic is their repeating spacers and direct repeats. This characteristic makes CRISPRs easily identifiable in long sequences of DNA, since the number of repeats decreases the likelihood of a false positive match.
Analysis of CRISPRs in metagenomic data is more challenging, as CRISPR loci do not typically assemble, due to their repetitive nature or through strain variation, which confuses assembly algorithms. Where many reference genomes are available, polymerase chain reaction (PCR) can be used to amplify CRISPR arrays and analyse spacer content. However, this approach yields information only for specifically targeted CRISPRs and for organisms with sufficient representation in public databases to design reliable polymerase chain reaction (PCR) primers. Degenerate repeat-specific primers can be used to amplify CRISPR spacers directly from environmental samples; amplicons containing two or three spacers can be then computationally assembled to reconstruct long CRISPR arrays.
The alternative is to extract and reconstruct CRISPR arrays from shotgun metagenomic data. This is computationally more difficult, particularly with second generation sequencing technologies (e.g. 454, Illumina), as the short read lengths prevent more than two or three repeat units appearing in a single read. CRISPR identification in raw reads has been achieved using purely de novo identification or by using direct repeat sequences in partially assembled CRISPR arrays from contigs (overlapping DNA segments that together represent a consensus region of DNA) and direct repeat sequences from published genomes as a hook for identifying direct repeats in individual reads.
Use by phages
Another way for bacteria to defend against phage infection is by having chromosomal islands. A subtype of chromosomal islands called phage-inducible chromosomal island (PICI) is excised from a bacterial chromosome upon phage infection and can inhibit phage replication. PICIs are induced, excised, replicated, and finally packaged into small capsids by certain staphylococcal temperate phages. PICIs use several mechanisms to block phage reproduction. In the first mechanism, PICI-encoded Ppi differentially blocks phage maturation by binding or interacting specifically with phage TerS, hence blocking phage TerS/TerL complex formation responsible for phage DNA packaging. In the second mechanism PICI CpmAB redirects the phage capsid morphogenetic protein to make 95% of SaPI-sized capsid and phage DNA can package only 1/3rd of their genome in these small capsids and hence become nonviable phage. The third mechanism involves two proteins, PtiA and PtiB, that target the LtrC, which is responsible for the production of virion and lysis proteins. This interference mechanism is modulated by a modulatory protein, PtiM, binds to one of the interference-mediating proteins, PtiA, and hence achieves the required level of interference.
One study showed that lytic ICP1 phage, which specifically targets Vibrio cholerae serogroup O1, has acquired a CRISPR/Cas system that targets a V. cholera PICI-like element. The system has 2 CRISPR loci and 9 Cas genes. It seems to be homologous to the I-F system found in Yersinia pestis. Moreover, like the bacterial CRISPR/Cas system, ICP1 CRISPR/Cas can acquire new sequences, which allows phage and host to co-evolve.
Certain archaeal viruses were shown to carry mini-CRISPR arrays containing one or two spacers. It has been shown that spacers within the virus-borne CRISPR arrays target other viruses and plasmids, suggesting that mini-CRISPR arrays represent a mechanism of heterotypic superinfection exclusion and participate in interviral conflicts.
Applications
CRISPR gene editing
CRISPR technology has been applied in the food and farming industries to engineer probiotic cultures and to immunize industrial cultures (for yogurt, for instance) against infections. It is also being used in crops to enhance yield, drought tolerance and nutritional value.
By the end of 2014, some 1000 research papers had been published that mentioned CRISPR. The technology had been used to functionally inactivate genes in human cell lines and cells, to study Candida albicans, to modify yeasts used to make biofuels, and genetically modify crop strains. Hsu and his colleagues state that the ability to manipulate the genetic sequences allows for reverse engineering that can positively affect biofuel production. CRISPR can also be used to change mosquitoes so they cannot transmit diseases such as malaria. CRISPR-based approaches utilizing Cas12a have recently been utilized in the successful modification of a broad number of plant species.
In July 2019, CRISPR was used to experimentally treat a patient with a genetic disorder. The patient was a 34-year-old woman with sickle cell disease.
In February 2020, progress was made on HIV treatments with 60-80% of the integrated viral DNA removed in mice and some being completely free from the virus after edits involving both LASER ART, a new anti-retroviral therapy, and CRISPR.
In March 2020, CRISPR-modified virus was injected into a patient's eye in an attempt to treat Leber congenital amaurosis.
In the future, CRISPR gene editing could potentially be used to create new species or revive extinct species from closely related ones.
CRISPR-based re-evaluations of claims for gene-disease relationships have led to the discovery of potentially important anomalies.
In July 2021, CRISPR gene editing of hiPSC's was used to study the role of MBNL proteins associated with DM1.
CRISPR as diagnostic tool

CRISPR associated nucleases have shown to be useful as a tool for molecular testing due to their ability to specifically target nucleic acid sequences in a high background of non-target sequences. In 2016, the Cas9 nuclease was used to deplete unwanted nucleotide sequences in next-generation sequencing libraries while requiring only 250 picograms of initial RNA input. Beginning in 2017, CRISPR associated nucleases were also used for direct diagnostic testing of nucleic acids, down to single molecule sensitivity. CRISPR diversity is used as an analysis target to discern phylogeny and diversity in bacteria, such as in xanthomonads by Martins et al., 2019. Early detections of plant pathogens by molecular typing of the pathogen's CRISPRs can be used in agriculture as demonstrated by Shen et al., 2020.
By coupling CRISPR-based diagnostics to additional enzymatic processes, the detection of molecules beyond nucleic acids is possible. One example of a coupled technology is SHERLOCK-based Profiling of IN vitro Transcription (SPRINT). SPRINT can be used to detect a variety of substances, such as metabolites in patient samples or contaminants in environmental samples, with high throughput or with portable point-of-care devices. CRISPR/Cas platforms are also being explored for detection and inactivation of SARS-CoV-2, the virus that causes COVID-19. Two different comprehensive diagnostic tests, AIOD-CRISPR and SHERLOCK test have been identified for SARS-CoV-2. The SHERLOCK test is based on a fluorescently labelled press reporter RNA which has the ability to identify 10 copies per microliter. The AIOD-CRISPR helps with robust and highly sensitive visual detection of the viral nucleic acid.
See also
Further reading
- Doudna J, Mali P (23 March 2016). CRISPR-Cas: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press. ISBN 978-1-62182-131-1.
- Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J (August 2016). "Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems" (PDF). Science. 353 (6299): aad5147. doi:10.1126/science.aad5147. hdl:1721.1/113195. PMID 27493190. S2CID 11086282.
- Sander JD, Joung JK (April 2014). "CRISPR-Cas systems for editing, regulating and targeting genomes". Nature Biotechnology. 32 (4): 347–355. doi:10.1038/nbt.2842. PMC 4022601. PMID 24584096.
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F (January 2016). "Rationally engineered Cas9 nucleases with improved specificity". Science. 351 (6268): 84–88. Bibcode:2016Sci...351...84S. doi:10.1126/science.aad5227. PMC 4714946. PMID 26628643.
- Terns RM, Terns MP (March 2014). "CRISPR-based technologies: prokaryotic defense weapons repurposed". Trends in Genetics. 30 (3): 111–118. doi:10.1016/j.tig.2014.01.003. PMC 3981743. PMID 24555991.
- Westra ER, Buckling A, Fineran PC (May 2014). "CRISPR-Cas systems: beyond adaptive immunity". Nature Reviews Microbiology. 12 (5): 317–326. doi:10.1038/nrmicro3241. PMID 24704746. S2CID 36575361.
- Andersson AF, Banfield JF (May 2008). "Virus population dynamics and acquired virus resistance in natural microbial communities". Science. 320 (5879): 1047–1050. Bibcode:2008Sci...320.1047A. doi:10.1126/science.1157358. PMID 18497291. S2CID 26209623.
- Hale C, Kleppe K, Terns RM, Terns MP (December 2008). "Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus". RNA. 14 (12): 2572–2579. doi:10.1261/rna.1246808. PMC 2590957. PMID 18971321.
- van der Ploeg JR (June 2009). "Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages". Microbiology. 155 (Pt 6): 1966–1976. doi:10.1099/mic.0.027508-0. PMID 19383692.
- van der Oost J, Brouns SJ (November 2009). "RNAi: prokaryotes get in on the act". Cell. 139 (5): 863–865. doi:10.1016/j.cell.2009.11.018. PMID 19945373. S2CID 11863610.
- Karginov FV, Hannon GJ (January 2010). "The CRISPR system: small RNA-guided defense in bacteria and archaea". Molecular Cell. 37 (1): 7–19. doi:10.1016/j.molcel.2009.12.033. PMC 2819186. PMID 20129051.
- Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R (March 2010). "Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS". Molecular Microbiology. 75 (6): 1495–1512. doi:10.1111/j.1365-2958.2010.07073.x. PMID 20132443. S2CID 37529215.
- Díez-Villaseñor C, Almendros C, García-Martínez J, Mojica FJ (May 2010). "Diversity of CRISPR loci in Escherichia coli". Microbiology. 156 (Pt 5): 1351–1361. doi:10.1099/mic.0.036046-0. PMID 20133361.
- Deveau H, Garneau JE, Moineau S (2010). "CRISPR/Cas system and its role in phage-bacteria interactions". Annual Review of Microbiology. 64: 475–493. doi:10.1146/annurev.micro.112408.134123. PMID 20528693.
- Koonin EV, Makarova KS (December 2009). "CRISPR-Cas: an adaptive immunity system in prokaryotes". F1000 Biology Reports. 1: 95. doi:10.3410/B1-95. PMC 2884157. PMID 20556198.
- "The age of the red pen". The Economist. August 22, 2015. ISSN 0013-0613. Retrieved 2015-08-25.
- Ran AF, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013). "Genome engineering using the CRISPR-Cas9 system". Nature Protocols. 8 (11): 2281–2308. doi:10.1038/nprot.2013.143. PMC 3969860. PMID 24157548.
External links
- "Advanced Gene Editing: CRISPR-Cas9" (PDF). Congressional Research Service.
- "Jennifer Doudna talk: Genome Engineering with CRISPR-Cas9: Birth of a Breakthrough Technology".
- "Human Nature". NOVA. Season 47. Episode 9. September 9, 2020. PBS. WGBH. Retrieved April 7, 2023.
Protein Data Bank
- Overview of all the structural information available in the PDB for UniProt: Q46901 (CRISPR system Cascade subunit CasA) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: P76632 (CRISPR system Cascade subunit CasB) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: Q46899 (CRISPR system Cascade subunit CasC) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: Q46898 (CRISPR system Cascade subunit CasD) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: Q46897 (CRISPR system Cascade subunit CasE) at the PDBe-KB.