
In molecular biology, an interactome is the whole set of molecular interactions in a particular cell. The term specifically refers to physical interactions among molecules (such as those among proteins, also known as protein–protein interactions, PPIs; or between small molecules and proteins) but can also describe sets of indirect interactions among genes (genetic interactions).
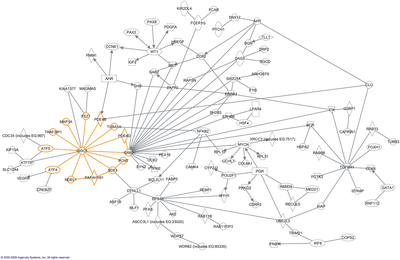
The word "interactome" was originally coined in 1999 by a group of French scientists headed by Bernard Jacq. Mathematically, interactomes are generally displayed as graphs. Though interactomes may be described as biological networks, they should not be confused with other networks such as neural networks or food webs.
Molecular interaction networks
Molecular interactions can occur between molecules belonging to different biochemical families (proteins, nucleic acids, lipids, carbohydrates, etc.) and also within a given family. Whenever such molecules are connected by physical interactions, they form molecular interaction networks that are generally classified by the nature of the compounds involved. Most commonly, interactome refers to protein–protein interaction (PPI) network (PIN) or subsets thereof. For instance, the Sirt-1 protein interactome and Sirt family second order interactome is the network involving Sirt-1 and its directly interacting proteins where as second order interactome illustrates interactions up to second order of neighbors (Neighbors of neighbors). Another extensively studied type of interactome is the protein–DNA interactome, also called a gene-regulatory network, a network formed by transcription factors, chromatin regulatory proteins, and their target genes. Even metabolic networks can be considered as molecular interaction networks: metabolites, i.e. chemical compounds in a cell, are converted into each other by enzymes, which have to bind their substrates physically.
In fact, all interactome types are interconnected. For instance, protein interactomes contain many enzymes which in turn form biochemical networks. Similarly, gene regulatory networks overlap substantially with protein interaction networks and signaling networks.
Size

It has been suggested that the size of an organism's interactome correlates better than genome size with the biological complexity of the organism. Although protein–protein interaction maps containing several thousand binary interactions are now available for several species, none of them is presently complete and the size of interactomes is still a matter of debate.
Yeast
The yeast interactome, i.e. all protein–protein interactions among proteins of Saccharomyces cerevisiae, has been estimated to contain between 10,000 and 30,000 interactions. A reasonable estimate may be on the order of 20,000 interactions. Larger estimates often include indirect or predicted interactions, often from affinity purification/mass spectrometry (AP/MS) studies.
Genetic interaction networks
Genes interact in the sense that they affect each other's function. For instance, a mutation may be harmless, but when it is combined with another mutation, the combination may turn out to be lethal. Such genes are said to "interact genetically". Genes that are connected in such a way form genetic interaction networks. Some of the goals of these networks are: develop a functional map of a cell's processes, drug target identification using chemoproteomics, and to predict the function of uncharacterized genes.
In 2010, the most "complete" gene interactome produced to date was compiled from about 5.4 million two-gene comparisons to describe "the interaction profiles for ~75% of all genes in the budding yeast", with ~170,000 gene interactions. The genes were grouped based on similar function so as to build a functional map of the cell's processes. Using this method the study was able to predict known gene functions better than any other genome-scale data set as well as adding functional information for genes that hadn't been previously described. From this model genetic interactions can be observed at multiple scales which will assist in the study of concepts such as gene conservation. Some of the observations made from this study are that there were twice as many negative as positive interactions, negative interactions were more informative than positive interactions, and genes with more connections were more likely to result in lethality when disrupted.
Interactomics
Interactomics is a discipline at the intersection of bioinformatics and biology that deals with studying both the interactions and the consequences of those interactions between and among proteins, and other molecules within a cell. Interactomics thus aims to compare such networks of interactions (i.e., interactomes) between and within species in order to find how the traits of such networks are either preserved or varied.
Interactomics is an example of "top-down" systems biology, which takes an overhead view of a biosystem or organism. Large sets of genome-wide and proteomic data are collected, and correlations between different molecules are inferred. From the data new hypotheses are formulated about feedbacks between these molecules. These hypotheses can then be tested by new experiments.
Experimental methods to map interactomes
The study of interactomes is called interactomics. The basic unit of a protein network is the protein–protein interaction (PPI). While there are numerous methods to study PPIs, there are relatively few that have been used on a large scale to map whole interactomes.
The yeast two hybrid system (Y2H) is suited to explore the binary interactions among two proteins at a time. Affinity purification and subsequent mass spectrometry is suited to identify a protein complex. Both methods can be used in a high-throughput (HTP) fashion. Yeast two hybrid screens allow false positive interactions between proteins that are never expressed in the same time and place; affinity capture mass spectrometry does not have this drawback, and is the current gold standard. Yeast two-hybrid data better indicates non-specific tendencies towards sticky interactions rather while affinity capture mass spectrometry better indicates functional in vivo protein–protein interactions.
Computational methods to study interactomes
Once an interactome has been created, there are numerous ways to analyze its properties. However, there are two important goals of such analyses. First, scientists try to elucidate the systems properties of interactomes, e.g. the topology of its interactions. Second, studies may focus on individual proteins and their role in the network. Such analyses are mainly carried out using bioinformatics methods and include the following, among many others:
Validation
First, the coverage and quality of an interactome has to be evaluated. Interactomes are never complete, given the limitations of experimental methods. For instance, it has been estimated that typical Y2H screens detect only 25% or so of all interactions in an interactome. The coverage of an interactome can be assessed by comparing it to benchmarks of well-known interactions that have been found and validated by independent assays. Other methods filter out false positives calculating the similarity of known annotations of the proteins involved or define a likelihood of interaction using the subcellular localization of these proteins.
Predicting PPIs
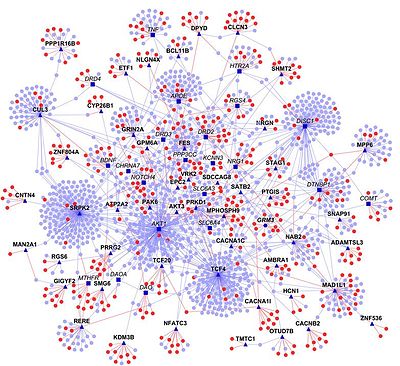
Using experimental data as a starting point, homology transfer is one way to predict interactomes. Here, PPIs from one organism are used to predict interactions among homologous proteins in another organism ("interologs"). However, this approach has certain limitations, primarily because the source data may not be reliable (e.g. contain false positives and false negatives). In addition, proteins and their interactions change during evolution and thus may have been lost or gained. Nevertheless, numerous interactomes have been predicted, e.g. that of Bacillus licheniformis.
Some algorithms use experimental evidence on structural complexes, the atomic details of binding interfaces and produce detailed atomic models of protein–protein complexes as well as other protein–molecule interactions. Other algorithms use only sequence information, thereby creating unbiased complete networks of interaction with many mistakes.
Some methods use machine learning to distinguish how interacting protein pairs differ from non-interacting protein pairs in terms of pairwise features such as cellular colocalization, gene co-expression, how closely located on a DNA are the genes that encode the two proteins, and so on.Random Forest has been found to be most-effective machine learning method for protein interaction prediction. Such methods have been applied for discovering protein interactions on human interactome, specifically the interactome of Membrane proteins and the interactome of Schizophrenia-associated proteins.
Text mining of PPIs
Some efforts have been made to extract systematically interaction networks directly from the scientific literature. Such approaches range in terms of complexity from simple co-occurrence statistics of entities that are mentioned together in the same context (e.g. sentence) to sophisticated natural language processing and machine learning methods for detecting interaction relationships.
Protein function prediction
Protein interaction networks have been used to predict the function of proteins of unknown functions. This is usually based on the assumption that uncharacterized proteins have similar functions as their interacting proteins (guilt by association). For example, YbeB, a protein of unknown function was found to interact with ribosomal proteins and later shown to be involved in bacterial and eukaryotic (but not archaeal) translation. Although such predictions may be based on single interactions, usually several interactions are found. Thus, the whole network of interactions can be used to predict protein functions, given that certain functions are usually enriched among the interactors. The term hypothome has been used to denote an interactome wherein at least one of the genes or proteins is a hypothetical protein.
Perturbations and disease
The topology of an interactome makes certain predictions how a network reacts to the perturbation (e.g. removal) of nodes (proteins) or edges (interactions). Such perturbations can be caused by mutations of genes, and thus their proteins, and a network reaction can manifest as a disease. A network analysis can identify drug targets and biomarkers of diseases.
Network structure and topology
Interaction networks can be analyzed using the tools of graph theory. Network properties include the degree distribution, clustering coefficients, betweenness centrality, and many others. The distribution of properties among the proteins of an interactome has revealed that the interactome networks often have scale-free topology where functional modules within a network indicate specialized subnetworks. Such modules can be functional, as in a signaling pathway, or structural, as in a protein complex. In fact, it is a formidable task to identify protein complexes in an interactome, given that a network on its own does not directly reveal the presence of a stable complex.
Studied interactomes
Viral interactomes
Viral protein interactomes consist of interactions among viral or phage proteins. They were among the first interactome projects as their genomes are small and all proteins can be analyzed with limited resources. Viral interactomes are connected to their host interactomes, forming virus-host interaction networks. Some published virus interactomes include
Bacteriophage
- Escherichia coli bacteriophage lambda
- Escherichia coli bacteriophage T7
- Streptococcus pneumoniae bacteriophage Dp-1
- Streptococcus pneumoniae bacteriophage Cp-1
The lambda and VZV interactomes are not only relevant for the biology of these viruses but also for technical reasons: they were the first interactomes that were mapped with multiple Y2H vectors, proving an improved strategy to investigate interactomes more completely than previous attempts have shown.
Human (mammalian) viruses
- Human varicella zoster virus (VZV)
- Chandipura virus
- Epstein-Barr virus (EBV)
- Hepatitis C virus (HPC), Human-HCV interactions
- Hepatitis E virus (HEV)
- Herpes simplex virus 1 (HSV-1)
- Kaposi's sarcoma-associated herpesvirus (KSHV)
- Murine cytomegalovirus (mCMV)
Bacterial interactomes
Relatively few bacteria have been comprehensively studied for their protein–protein interactions. However, none of these interactomes are complete in the sense that they captured all interactions. In fact, it has been estimated that none of them covers more than 20% or 30% of all interactions, primarily because most of these studies have only employed a single method, all of which discover only a subset of interactions. Among the published bacterial interactomes (including partial ones) are
| Species | proteins total | interactions | type | reference |
| Helicobacter pylori | 1,553 | ~3,004 | Y2H | |
| Campylobacter jejuni | 1,623 | 11,687 | Y2H | |
| Treponema pallidum | 1,040 | 3,649 | Y2H | |
| Escherichia coli | 4,288 | (5,993) | AP/MS | |
| Escherichia coli | 4,288 | 2,234 | Y2H | |
| Mesorhizobium loti | 6,752 | 3,121 | Y2H | |
| Mycobacterium tuberculosis | 3,959 | >8000 | B2H | |
| Mycoplasma genitalium | 482 | AP/MS | ||
| Synechocystis sp. PCC6803 | 3,264 | 3,236 | Y2H | |
| Staphylococcus aureus (MRSA) | 2,656 | 13,219 | AP/MS |
The E. coli and Mycoplasma interactomes have been analyzed using large-scale protein complex affinity purification and mass spectrometry (AP/MS), hence it is not easily possible to infer direct interactions. The others have used extensive yeast two-hybrid (Y2H) screens. The Mycobacterium tuberculosis interactome has been analyzed using a bacterial two-hybrid screen (B2H).
Note that numerous additional interactomes have been predicted using computational methods (see section above).
Eukaryotic interactomes
There have been several efforts to map eukaryotic interactomes through HTP methods. While no biological interactomes have been fully characterized, over 90% of proteins in Saccharomyces cerevisiae have been screened and their interactions characterized, making it the best-characterized interactome. Species whose interactomes have been studied in some detail include
Recently, the pathogen-host interactomes of Hepatitis C Virus/Human (2008), Epstein Barr virus/Human (2008), Influenza virus/Human (2009) were delineated through HTP to identify essential molecular components for pathogens and for their host's immune system.
Predicted interactomes
As described above, PPIs and thus whole interactomes can be predicted. While the reliability of these predictions is debatable, they are providing hypotheses that can be tested experimentally. Interactomes have been predicted for a number of species, e.g.
- Human (Homo sapiens)
- Rice (Oryza sativa)
- Xanthomonas oryzae
- Arabidopsis thaliana
- Tomato (Solanum lycopersicum)
- Field mustard (Brassica rapa)
- Maize, corn (Zea mays)
- Poplar (Populus trichocarpa)
- SARS-CoV-2

Network properties
Protein interaction networks can be analyzed with the same tool as other networks. In fact, they share many properties with biological or social networks. Some of the main characteristics are as follows.

Degree distribution
The degree distribution describes the number of proteins that have a certain number of connections. Most protein interaction networks show a scale-free (power law) degree distribution where the connectivity distribution P(k) ~ k−γ with k being the degree. This relationship can also be seen as a straight line on a log-log plot since, the above equation is equal to log(P(k)) ~ —y•log(k). One characteristic of such distributions is that there are many proteins with few interactions and few proteins that have many interactions, the latter being called "hubs".
Hubs
Highly connected nodes (proteins) are called hubs. Han et al. have coined the term "party hub" for hubs whose expression is correlated with its interaction partners. Party hubs also connect proteins within functional modules such as protein complexes. In contrast, "date hubs" do not exhibit such a correlation and appear to connect different functional modules. Party hubs are found predominantly in AP/MS data sets, whereas date hubs are found predominantly in binary interactome network maps. Note that the validity of the date hub/party hub distinction was disputed. Party hubs generally consist of multi-interface proteins whereas date hubs are more frequently single-interaction interface proteins. Consistent with a role for date-hubs in connecting different processes, in yeast the number of binary interactions of a given protein is correlated to the number of phenotypes observed for the corresponding mutant gene in different physiological conditions.
Modules
Nodes involved in the same biochemical process are highly interconnected.
Evolution
The evolution of interactome complexity is delineated in a study published in Nature. In this study it is first noted that the boundaries between prokaryotes, unicellular eukaryotes and multicellular eukaryotes are accompanied by orders-of-magnitude reductions in effective population size, with concurrent amplifications of the effects of random genetic drift. The resultant decline in the efficiency of selection seems to be sufficient to influence a wide range of attributes at the genomic level in a nonadaptive manner. The Nature study shows that the variation in the power of random genetic drift is also capable of influencing phylogenetic diversity at the subcellular and cellular levels. Thus, population size would have to be considered as a potential determinant of the mechanistic pathways underlying long-term phenotypic evolution. In the study it is further shown that a phylogenetically broad inverse relation exists between the power of drift and the structural integrity of protein subunits. Thus, the accumulation of mildly deleterious mutations in populations of small size induces secondary selection for protein–protein interactions that stabilize key gene functions, mitigating the structural degradation promoted by inefficient selection. By this means, the complex protein architectures and interactions essential to the genesis of phenotypic diversity may initially emerge by non-adaptive mechanisms.
Criticisms, challenges, and responses
Kiemer and Cesareni raise the following concerns with the state (circa 2007) of the field especially with the comparative interactomic: The experimental procedures associated with the field are error prone leading to "noisy results". This leads to 30% of all reported interactions being artifacts. In fact, two groups using the same techniques on the same organism found less than 30% interactions in common. However, some authors have argued that such non-reproducibility results from the extraordinary sensitivity of various methods to small experimental variation. For instance, identical conditions in Y2H assays result in very different interactions when different Y2H vectors are used.
Techniques may be biased, i.e. the technique determines which interactions are found. In fact, any method has built in biases, especially protein methods. Because every protein is different no method can capture the properties of each protein. For instance, most analytical methods that work fine with soluble proteins deal poorly with membrane proteins. This is also true for Y2H and AP/MS technologies.
Interactomes are not nearly complete with perhaps the exception of S. cerevisiae. This is not really a criticism as any scientific area is "incomplete" initially until the methodologies have been improved. Interactomics in 2015 is where genome sequencing was in the late 1990s, given that only a few interactome datasets are available (see table above).
While genomes are stable, interactomes may vary between tissues, cell types, and developmental stages. Again, this is not a criticism, but rather a description of the challenges in the field.
It is difficult to match evolutionarily related proteins in distantly related species. While homologous DNA sequences can be found relatively easily, it is much more difficult to predict homologous interactions ("interologs") because the homologs of two interacting proteins do not need to interact. For instance, even within a proteome two proteins may interact but their paralogs may not.
Each protein–protein interactome may represent only a partial sample of potential interactions, even when a supposedly definitive version is published in a scientific journal. Additional factors may have roles in protein interactions that have yet to be incorporated in interactomes. The binding strength of the various protein interactors, microenvironmental factors, sensitivity to various procedures, and the physiological state of the cell all impact protein–protein interactions, yet are usually not accounted for in interactome studies.
See also
- Bioinformatics, Omics, Proteomics, Genomics
- Biological networks
- BioPlex
- Connectome
- Glossary of graph theory
- Human interactome
- Interaction network
- List of omics topics in biology
- Mathematical biology
- Metabolic network
- Metabolic network modelling
- Metabolic pathway
- Network medicine
- Protein–protein interactions
- Systems biology
- Chemoproteomics
Further reading
- Park J, Lappe M, Teichmann SA (Mar 2001). "Mapping protein family interactions: intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast". J Mol Biol. 307 (3): 929–38. doi:10.1006/jmbi.2001.4526. PMID 11273711.
External links
Interactome web servers
- Protinfo PPC predicts the atomic 3D structure of protein protein complexes.Kittichotirat W, Guerquin M, Bumgarner R, Samudrala R (2009). "Protinfo PPC: A web server for atomic level prediction of protein complexes". Nucleic Acids Research. 37 (Web Server issue): W519–W525. doi:10.1093/nar/gkp306. PMC 2703994. PMID 19420059.
- IBIS (server) reports, predicts and integrates multiple types of conserved interactions for proteins.
Interactome visualization tools
- GPS-Prot Web-based data visualization for protein interactions
- PINV - Protein Interaction Network Visualizer
Interactome databases
- BioGRID database
- mentha the interactome browser Calderone; et al. (2013). "mentha: a resource for browsing integrated protein-interaction networks". Nature Methods. 10 (8): 690–691. doi:10.1038/nmeth.2561. PMID 23900247. S2CID 9733108.
- IntAct: The Molecular Interaction Database
- Interactome.org — a dedicated interactome web site.