

The pharmaceutical industry discovers, develops, produces, and markets drugs or pharmaceuticals for use as medications to be administered to patients (or self-administered), with the aim to mitigate symptoms or disease conditions, vaccinate them, or alleviate symptoms of disease or surgical or disease condition.Pharmaceutical companies may deal in generic or brand medications and medical devices. They are subject to a variety of laws and regulations that govern the patenting, testing, safety, efficacy using drug testing and marketing of drugs. The global pharmaceuticals market produced treatments worth $1,228.45 billion in 2020 and showed a compound annual growth rate (CAGR) of 1.8%.
History
Mid-1800s – 1945: From botanicals to the first synthetic drugs
The modern era of pharmaceutical industry began with local apothecaries that expanded from their traditional role of distributing botanical drugs such as morphine and quinine to wholesale manufacture in the mid-1800s, and from discoveries resulting from applied research. Intentional drug discovery from plants began with the isolation between 1803 and 1805 of morphine - an analgesic and sleep-inducing agent - from opium by the German apothecary assistant Friedrich Sertürner, who named this compound after the Greek god of dreams, Morpheus. By the late 1880s, German dye manufacturers had perfected the purification of individual organic compounds from tar and other mineral sources and had also established rudimentary methods in organic chemical synthesis. The development of synthetic chemical methods allowed scientists to systematically vary the structure of chemical substances, and growth in the emerging science of pharmacology expanded their ability to evaluate the biological effects of these structural changes.
Epinephrine, norepinephrine, and amphetamines
By the 1890s, the profound effect of adrenal extracts on many different tissue types had been discovered, setting off a search both for the mechanism of chemical signaling and efforts to exploit these observations for the development of new drugs. The blood pressure raising and vasoconstrictive effects of adrenal extracts were of particular interest to surgeons as hemostatic agents and as treatment for shock, and a number of companies developed products based on adrenal extracts containing varying purities of the active substance. In 1897, John Abel of Johns Hopkins University identified the active principle as epinephrine, which he isolated in an impure state as the sulfate salt. Industrial chemist Jōkichi Takamine later developed a method for obtaining epinephrine in a pure state, and licensed the technology to Parke-Davis. Parke-Davis marketed epinephrine under the trade name Adrenalin. Injected epinephrine proved to be especially efficacious for the acute treatment of asthma attacks, and an inhaled version was sold in the United States until 2011 (Primatene Mist). By 1929 epinephrine had been formulated into an inhaler for use in the treatment of nasal congestion.
While highly effective, the requirement for injection limited the use of epinephrine and orally active derivatives were sought. A structurally similar compound, ephedrine, was identified by Japanese chemists in the Ma Huang plant and marketed by Eli Lilly as an oral treatment for asthma. Following the work of Henry Dale and George Barger at Burroughs-Wellcome, academic chemist Gordon Alles synthesized amphetamine and tested it in asthma patients in 1929. The drug proved to have only modest anti-asthma effects but produced sensations of exhilaration and palpitations. Amphetamine was developed by Smith, Kline and French as a nasal decongestant under the trade name Benzedrine Inhaler. Amphetamine was eventually developed for the treatment of narcolepsy, post-encephalitic parkinsonism, and mood elevation in depression and other psychiatric indications. It received approval as a New and Nonofficial Remedy from the American Medical Association for these uses in 1937, and remained in common use for depression until the development of tricyclic antidepressants in the 1960s.
Discovery and development of the barbiturate

In 1903, Hermann Emil Fischer and Joseph von Mering disclosed their discovery that diethylbarbituric acid, formed from the reaction of diethylmalonic acid, phosphorus oxychloride and urea, induces sleep in dogs. The discovery was patented and licensed to Bayer pharmaceuticals, which marketed the compound under the trade name Veronal as a sleep aid beginning in 1904. Systematic investigations of the effect of structural changes on potency and duration of action led to the discovery of phenobarbital at Bayer in 1911 and the discovery of its potent anti-epileptic activity in 1912. Phenobarbital was among the most widely used drugs for the treatment of epilepsy through the 1970s, and as of 2014, remains on the World Health Organizations list of essential medications. The 1950s and 1960s saw increased awareness of the addictive properties and abuse potential of barbiturates and amphetamines and led to increasing restrictions on their use and growing government oversight of prescribers. Today, amphetamine is largely restricted to use in the treatment of attention deficit disorder and phenobarbital in the treatment of epilepsy.
In 1958, Leo Sternbach discovered the first benzodiazepine, chlordiazepoxide (Librium). Dozens of other benzodiazepines have been developed and are in use, some of the more popular drugs being diazepam (Valium), alprazolam (Xanax), clonazepam (Klonopin), and lorazepam (Ativan). Due to their far superior safety and therapeutic properties, benzodiazepines have largely replaced the use of barbiturates in medicine, except in certain special cases. When it was later discovered that benzodiazepines, like barbiturates, significantly lose their effectiveness and can have serious side effects when taken long-term, Heather Ashton researched benzodiazepine dependence and developed a protocol to discontinue their use.
Insulin
A series of experiments performed from the late 1800s to the early 1900s revealed that diabetes is caused by the absence of a substance normally produced by the pancreas. In 1869, Oskar Minkowski and Joseph von Mering found that diabetes could be induced in dogs by surgical removal of the pancreas. In 1921, Canadian professor Frederick Banting and his student Charles Best repeated this study and found that injections of pancreatic extract reversed the symptoms produced by pancreas removal. Soon, the extract was demonstrated to work in people, but development of insulin therapy as a routine medical procedure was delayed by difficulties in producing the material in sufficient quantity and with reproducible purity. The researchers sought assistance from industrial collaborators at Eli Lilly and Co. based on the company's experience with large scale purification of biological materials. Chemist George B. Walden of Eli Lilly and Company found that careful adjustment of the pH of the extract allowed a relatively pure grade of insulin to be produced. Under pressure from Toronto University and a potential patent challenge by academic scientists who had independently developed a similar purification method, an agreement was reached for non-exclusive production of insulin by multiple companies. Prior to the discovery and widespread availability of insulin therapy the life expectancy of diabetics was only a few months.
Early anti-infective research: Salvarsan, Prontosil, Penicillin and vaccines
The development of drugs for the treatment of infectious diseases was a major focus of early research and development efforts; in 1900, pneumonia, tuberculosis, and diarrhea were the three leading causes of death in the United States and mortality in the first year of life exceeded 10%.
In 1911 arsphenamine, the first synthetic anti-infective drug, was developed by Paul Ehrlich and chemist Alfred Bertheim of the Institute of Experimental Therapy in Berlin. The drug was given the commercial name Salvarsan. Ehrlich, noting both the general toxicity of arsenic and the selective absorption of certain dyes by bacteria, hypothesized that an arsenic-containing dye with similar selective absorption properties could be used to treat bacterial infections. Arsphenamine was prepared as part of a campaign to synthesize a series of such compounds and found to exhibit partially selective toxicity. Arsphenamine proved to be the first effective treatment for syphilis, a disease which prior to that time was incurable and led inexorably to severe skin ulceration, neurological damage, and death.
Ehrlich's approach of systematically varying the chemical structure of synthetic compounds and measuring the effects of these changes on biological activity was pursued broadly by industrial scientists, including Bayer scientists Josef Klarer, Fritz Mietzsch, and Gerhard Domagk. This work, also based in the testing of compounds available from the German dye industry, led to the development of Prontosil, the first representative of the sulfonamide class of antibiotics. Compared to arsphenamine, the sulfonamides had a broader spectrum of activity and were far less toxic, rendering them useful for infections caused by pathogens such as streptococci. In 1939, Domagk received the Nobel Prize in Medicine for this discovery. Nonetheless, the dramatic decrease in deaths from infectious diseases that occurred prior to World War II was primarily the result of improved public health measures such as clean water and less crowded housing, and the impact of anti-infective drugs and vaccines was significant mainly after World War II.
In 1928, Alexander Fleming discovered the antibacterial effects of penicillin, but its exploitation for the treatment of human disease awaited the development of methods for its large scale production and purification. These were developed by a U.S. and British government-led consortium of pharmaceutical companies during the Second World War.
Early progress toward the development of vaccines occurred throughout this period, primarily in the form of academic and government-funded basic research directed toward the identification of the pathogens responsible for common communicable diseases. In 1885, Louis Pasteur and Pierre Paul Émile Roux created the first rabies vaccine. The first diphtheria vaccines were produced in 1914 from a mixture of diphtheria toxin and antitoxin (produced from the serum of an inoculated animal), but the safety of the inoculation was marginal and it was not widely used. The United States recorded 206,000 cases of diphtheria in 1921 resulting in 15,520 deaths. In 1923, parallel efforts by Gaston Ramon at the Pasteur Institute and Alexander Glenny at the Wellcome Research Laboratories (later part of GlaxoSmithKline) led to the discovery that a safer vaccine could be produced by treating diphtheria toxin with formaldehyde. In 1944, Maurice Hilleman of Squibb Pharmaceuticals developed the first vaccine against Japanese Encephalitis. Hilleman would later move to Merck where he would play a key role in the development of vaccines against measles, mumps, chickenpox, rubella, hepatitis A, hepatitis B, and meningitis.
Unsafe drugs and early industry regulation

Prior to the 20th century, drugs were generally produced by small scale manufacturers with little regulatory control over manufacturing or claims of safety and efficacy. To the extent that such laws did exist, enforcement was lax. In the United States, increased regulation of vaccines and other biological drugs was spurred by tetanus outbreaks and deaths caused by the distribution of contaminated smallpox vaccine and diphtheria antitoxin. The Biologics Control Act of 1902 required that federal government grant premarket approval for every biological drug and for the process and facility producing such drugs. This was followed in 1906 by the Pure Food and Drugs Act, which forbade the interstate distribution of adulterated or misbranded foods and drugs. A drug was considered misbranded if it contained alcohol, morphine, opium, cocaine, or any of several other potentially dangerous or addictive drugs, and if its label failed to indicate the quantity or proportion of such drugs. The government's attempts to use the law to prosecute manufacturers for making unsupported claims of efficacy were undercut by a Supreme Court ruling restricting the federal government's enforcement powers to cases of incorrect specification of the drug's ingredients.
In 1937 over 100 people died after ingesting "Elixir Sulfanilamide" manufactured by S.E. Massengill Company of Tennessee. The product was formulated in diethylene glycol, a highly toxic solvent that is now widely used as antifreeze. Under the laws extant at that time, prosecution of the manufacturer was possible only under the technicality that the product had been called an "elixir", which literally implied a solution in ethanol. In response to this episode, the U.S. Congress passed the Federal Food, Drug, and Cosmetic Act of 1938, which for the first time required pre-market demonstration of safety before a drug could be sold, and explicitly prohibited false therapeutic claims.
The post-war years, 1945–1970
Further advances in anti-infective research
The aftermath of World War II saw an explosion in the discovery of new classes of antibacterial drugs including the cephalosporins (developed by Eli Lilly based on the seminal work of Giuseppe Brotzu and Edward Abraham),streptomycin (discovered during a Merck-funded research program in Selman Waksman's laboratory), the tetracyclines (discovered at Lederle Laboratories, now a part of Pfizer), erythromycin (discovered at Eli Lilly and Co.) and their extension to an increasingly wide range of bacterial pathogens. Streptomycin, discovered during a Merck-funded research program in Selman Waksman's laboratory at Rutgers in 1943, became the first effective treatment for tuberculosis. At the time of its discovery, sanitoriums for the isolation of tuberculosis-infected people were an ubiquitous feature of cities in developed countries, with 50% dying within 5 years of admission.
A Federal Trade Commission report issued in 1958 attempted to quantify the effect of antibiotic development on American public health. The report found that over the period 1946–1955, there was a 42% drop in the incidence of diseases for which antibiotics were effective and only a 20% drop in those for which antibiotics were not effective. The report concluded that "it appears that the use of antibiotics, early diagnosis, and other factors have limited the epidemic spread and thus the number of these diseases which have occurred". The study further examined mortality rates for eight common diseases for which antibiotics offered effective therapy (syphilis, tuberculosis, dysentery, scarlet fever, whooping cough, meningococcal infections, and pneumonia), and found a 56% decline over the same period. Notable among these was a 75% decline in deaths due to tuberculosis.
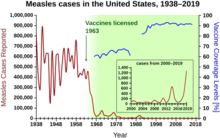
During the years 1940–1955, the rate of decline in the U.S. death rate accelerated from 2% per year to 8% per year, then returned to the historical rate of 2% per year. The dramatic decline in the immediate post-war years has been attributed to the rapid development of new treatments and vaccines for infectious disease that occurred during these years. Vaccine development continued to accelerate, with the most notable achievement of the period being Jonas Salk's 1954 development of the polio vaccine under the funding of the non-profit National Foundation for Infantile Paralysis. The vaccine process was never patented but was instead given to pharmaceutical companies to manufacture as a low-cost generic. In 1960 Maurice Hilleman of Merck Sharp & Dohme identified the SV40 virus, which was later shown to cause tumors in many mammalian species. It was later determined that SV40 was present as a contaminant in polio vaccine lots that had been administered to 90% of the children in the United States. The contamination appears to have originated both in the original cell stock and in monkey tissue used for production. In 2004 the United States Cancer Institute announced that it had concluded that SV40 is not associated with cancer in people.
Other notable new vaccines of the period include those for measles (1962, John Franklin Enders of Children's Medical Center Boston, later refined by Maurice Hilleman at Merck), Rubella (1969, Hilleman, Merck) and mumps (1967, Hilleman, Merck) The United States incidences of rubella, congenital rubella syndrome, measles, and mumps all fell by >95% in the immediate aftermath of widespread vaccination. The first 20 years of licensed measles vaccination in the U.S. prevented an estimated 52 million cases of the disease, 17,400 cases of mental retardation, and 5,200 deaths.
Development and marketing of antihypertensive drugs
Hypertension is a risk factor for atherosclerosis,heart failure,coronary artery disease,stroke,renal disease, and peripheral arterial disease, and is the most important risk factor for cardiovascular morbidity and mortality, in industrialized countries. Prior to 1940 approximately 23% of all deaths among persons over age 50 were attributed to hypertension. Severe cases of hypertension were treated by surgery.
Early developments in the field of treating hypertension included quaternary ammonium ion sympathetic nervous system blocking agents, but these compounds were never widely used due to their severe side effects, because the long-term health consequences of high blood pressure had not yet been established, and because they had to be administered by injection.
In 1952 researchers at Ciba discovered the first orally available vasodilator, hydralazine. A major shortcoming of hydralazine monotherapy was that it lost its effectiveness over time (tachyphylaxis). In the mid-1950s Karl H. Beyer, James M. Sprague, John E. Baer, and Frederick C. Novello of Merck and Co. discovered and developed chlorothiazide, which remains the most widely used antihypertensive drug today. This development was associated with a substantial decline in the mortality rate among people with hypertension. The inventors were recognized by a Public Health Lasker Award in 1975 for "the saving of untold thousands of lives and the alleviation of the suffering of millions of victims of hypertension".
A 2009 Cochrane review concluded that thiazide antihypertensive drugs reduce the risk of death (RR 0.89), stroke (RR 0.63), coronary heart disease (RR 0.84), and cardiovascular events (RR 0.70) in people with high blood pressure. In the ensuring years other classes of antihypertensive drug were developed and found wide acceptance in combination therapy, including loop diuretics (Lasix/furosemide, Hoechst Pharmaceuticals, 1963),beta blockers (ICI Pharmaceuticals, 1964)ACE inhibitors, and angiotensin receptor blockers. ACE inhibitors reduce the risk of new onset kidney disease [RR 0.71] and death [RR 0.84] in diabetic patients, irrespective of whether they have hypertension.
Oral Contraceptives
Prior to the second world war, birth control was prohibited in many countries, and in the United States even the discussion of contraceptive methods sometimes led to prosecution under Comstock laws. The history of the development of oral contraceptives is thus closely tied to the birth control movement and the efforts of activists Margaret Sanger, Mary Dennett, and Emma Goldman. Based on fundamental research performed by Gregory Pincus and synthetic methods for progesterone developed by Carl Djerassi at Syntex and by Frank Colton at G.D. Searle & Co., the first oral contraceptive, Enovid, was developed by E.D. Searle and Co. and approved by the FDA in 1960. The original formulation incorporated vastly excessive doses of hormones, and caused severe side effects. Nonetheless, by 1962, 1.2 million American women were on the pill, and by 1965 the number had increased to 6.5 million. The availability of a convenient form of temporary contraceptive led to dramatic changes in social mores including expanding the range of lifestyle options available to women, reducing the reliance of women on men for contraceptive practice, encouraging the delay of marriage, and increasing pre-marital co-habitation.
Thalidomide and the Kefauver-Harris Amendments

In the U.S., a push for revisions of the FD&C Act emerged from Congressional hearings led by Senator Estes Kefauver of Tennessee in 1959. The hearings covered a wide range of policy issues, including advertising abuses, questionable efficacy of drugs, and the need for greater regulation of the industry. While momentum for new legislation temporarily flagged under extended debate, a new tragedy emerged that underscored the need for more comprehensive regulation and provided the driving force for the passage of new laws.
On 12 September 1960, an American licensee, the William S. Merrell Company of Cincinnati, submitted a new drug application for Kevadon (thalidomide), a sedative that had been marketed in Europe since 1956. The FDA medical officer in charge of reviewing the compound, Frances Kelsey, believed that the data supporting the safety of thalidomide was incomplete. The firm continued to pressure Kelsey and the FDA to approve the application until November 1961, when the drug was pulled off the German market because of its association with grave congenital abnormalities. Several thousand newborns in Europe and elsewhere suffered the teratogenic effects of thalidomide. Without approval from the FDA, the firm distributed Kevadon to over 1,000 physicians there under the guise of investigational use. Over 20,000 Americans received thalidomide in this "study," including 624 pregnant patients, and about 17 known newborns suffered the effects of the drug.
The thalidomide tragedy resurrected Kefauver's bill to enhance drug regulation that had stalled in Congress, and the Kefauver-Harris Amendment became law on 10 October 1962. Manufacturers henceforth had to prove to FDA that their drugs were effective as well as safe before they could go on the US market. The FDA received authority to regulate advertising of prescription drugs and to establish good manufacturing practices. The law required that all drugs introduced between 1938 and 1962 had to be effective. An FDA - National Academy of Sciences collaborative study showed that nearly 40 percent of these products were not effective. A similarly comprehensive study of over-the-counter products began ten years later.
1970–1990s
Statins
In 1971, Akira Endo, a Japanese biochemist working for the pharmaceutical company Sankyo, identified mevastatin (ML-236B), a molecule produced by the fungus Penicillium citrinum, as an inhibitor of HMG-CoA reductase, a critical enzyme used by the body to produce cholesterol. Animal trials showed very good inhibitory effect as in clinical trials, however a long-term study in dogs found toxic effects at higher doses and as a result mevastatin was believed to be too toxic for human use. Mevastatin was never marketed, because of its adverse effects of tumors, muscle deterioration, and sometimes death in laboratory dogs. Statins above daily dose of 80 mg can cause serious muscle injury and rhabdomyolysis.
P. Roy Vagelos, chief scientist and later CEO of Merck & Co, was interested, and made several trips to Japan starting in 1975. By 1978, Merck had isolated lovastatin (mevinolin, MK803) from the fungus Aspergillus terreus, first marketed in 1987 as Mevacor.
In April 1994, the results of a Merck-sponsored study, the Scandinavian Simvastatin Survival Study, were announced. Researchers tested simvastatin, later sold by Merck as Zocor, on 4,444 patients with high cholesterol and heart disease. After five years, the study concluded the patients saw a 35% reduction in their cholesterol, and their chances of dying of a heart attack were reduced by 42%. In 1995, Zocor and Mevacor both made Merck over US$1 billion. Endo was awarded the 2006 Japan Prize, and the Lasker-DeBakey Clinical Medical Research Award in 2008. For his "pioneering research into a new class of molecules" for "lowering cholesterol,"
21st Century
Since several decades, biologics have been rising in importance in comparison with small molecules treatments. The biotech subsector, animal health and the Chinese pharmaceutical sector have also grown substantially. On the organizational side, big international pharma corporations have experienced a substantial decline of their value share. Also, the core generic sector (substitutions for off-patent brands) has been downvalued due to competition.
Torreya estimated the pharmaceutical industry to have a market valuation of USD 7.03 trillion by February 2021 from which USD 6.1 trillion is the value of the publicly-traded companies. Small Molecules modality had 58.2% of the valuation share down from 84.6% in 2003. Biologics was up at 30.5% from 14.5%. The valuation share of chinese Pharma grew from 2003 to 2021 from 1% to 12% overtaking Switzerland who is now ranked number 3 with 7.7%. The United States had still by far the most valued pharmaceutical industry with 40% of global valuation.
Impact of Mergers and Acquisitions
A 2022 article articulated this notion succinctly by saying "In the business of drug development, deals can be just as important as scientific breakthroughs", typically referred to as pharmaceutical M&A (for mergers and acquisitions). It highlighted that some of the most impactful of the remedies of the early 21st Century were only made possible through M&A activities, specifically noting Keytruda and Humira.
Research and development
Drug discovery is the process by which potential drugs are discovered or designed. In the past, most drugs have been discovered either by isolating the active ingredient from traditional remedies or by serendipitous discovery. Modern biotechnology often focuses on understanding the metabolic pathways related to a disease state or pathogen, and manipulating these pathways using molecular biology or biochemistry. A great deal of early-stage drug discovery has traditionally been carried out by universities and research institutions.
Drug development refers to activities undertaken after a compound is identified as a potential drug in order to establish its suitability as a medication. Objectives of drug development are to determine appropriate formulation and dosing, as well as to establish safety. Research in these areas generally includes a combination of in vitro studies, in vivo studies, and clinical trials. The cost of late stage development has meant it is usually done by the larger pharmaceutical companies. The pharmaceuticals and biotechnology industry spends more than 15% of its net sales for Research & Development which is in comparison with other industries by far the highest share.
Often, large multinational corporations exhibit vertical integration, participating in a broad range of drug discovery and development, manufacturing and quality control, marketing, sales, and distribution. Smaller organizations, on the other hand, often focus on a specific aspect such as discovering drug candidates or developing formulations. Often, collaborative agreements between research organizations and large pharmaceutical companies are formed to explore the potential of new drug substances. More recently, multi-nationals are increasingly relying on contract research organizations to manage drug development.
The cost of innovation
Drug discovery and development are very expensive; of all compounds investigated for use in humans only a small fraction are eventually approved in most nations by government-appointed medical institutions or boards, who have to approve new drugs before they can be marketed in those countries. In 2010 18 NMEs (New Molecular Entities) were approved and three biologics by the FDA, or 21 in total, which is down from 26 in 2009 and 24 in 2008. On the other hand, there were only 18 approvals in total in 2007 and 22 back in 2006. Since 2001, the Center for Drug Evaluation and Research has averaged 22.9 approvals a year. This approval comes only after heavy investment in pre-clinical development and clinical trials, as well as a commitment to ongoing safety monitoring. Drugs which fail part-way through this process often incur large costs, while generating no revenue in return. If the cost of these failed drugs is taken into account, the cost of developing a successful new drug (new chemical entity, or NCE), has been estimated at US$1.3 billion (not including marketing expenses). Professors Light and Lexchin reported in 2012, however, that the rate of approval for new drugs has been a relatively stable average rate of 15 to 25 for decades.
Industry-wide research and investment reached a record $65.3 billion in 2009. While the cost of research in the U.S. was about $34.2 billion between 1995 and 2010, revenues rose faster (revenues rose by $200.4 billion in that time).
A study by the consulting firm Bain & Company reported that the cost for discovering, developing and launching (which factored in marketing and other business expenses) a new drug (along with the prospective drugs that fail) rose over a five-year period to nearly $1.7 billion in 2003. According to Forbes, by 2010 development costs were between $4 billion to $11 billion per drug.
Some of these estimates also take into account the opportunity cost of investing capital many years before revenues are realized (see Time-value of money). Because of the very long time needed for discovery, development, and approval of pharmaceuticals, these costs can accumulate to nearly half the total expense. A direct consequence within the pharmaceutical industry value chain is that major pharmaceutical multinationals tend to increasingly outsource risks related to fundamental research, which somewhat reshapes the industry ecosystem with biotechnology companies playing an increasingly important role, and overall strategies being redefined accordingly. Some approved drugs, such as those based on re-formulation of an existing active ingredient (also referred to as Line-extensions) are much less expensive to develop.
Product approval
In the United States, new pharmaceutical products must be approved by the Food and Drug Administration (FDA) as being both safe and effective. This process generally involves submission of an Investigational New Drug filing with sufficient pre-clinical data to support proceeding with human trials. Following IND approval, three phases of progressively larger human clinical trials may be conducted. Phase I generally studies toxicity using healthy volunteers. Phase II can include pharmacokinetics and dosing in patients, and Phase III is a very large study of efficacy in the intended patient population. Following the successful completion of phase III testing, a New Drug Application is submitted to the FDA. The FDA reviews the data and if the product is seen as having a positive benefit-risk assessment, approval to market the product in the US is granted.
A fourth phase of post-approval surveillance is also often required due to the fact that even the largest clinical trials cannot effectively predict the prevalence of rare side-effects. Postmarketing surveillance ensures that after marketing the safety of a drug is monitored closely. In certain instances, its indication may need to be limited to particular patient groups, and in others the substance is withdrawn from the market completely.
The FDA provides information about approved drugs at the Orange Book site.
In the UK, the Medicines and Healthcare products Regulatory Agency approves and evaluates drugs for use. Normally an approval in the UK and other European countries comes later than one in the USA. Then it is the National Institute for Health and Care Excellence (NICE), for England and Wales, who decides if and how the National Health Service (NHS) will allow (in the sense of paying for) their use. The British National Formulary is the core guide for pharmacists and clinicians.
In many non-US western countries, a 'fourth hurdle' of cost effectiveness analysis has developed before new technologies can be provided. This focuses on the 'efficacy price tag' (in terms of, for example, the cost per QALY) of the technologies in question. In England and Wales NICE decides whether and in what circumstances drugs and technologies will be made available by the NHS, whilst similar arrangements exist with the Scottish Medicines Consortium in Scotland, and the Pharmaceutical Benefits Advisory Committee in Australia. A product must pass the threshold for cost-effectiveness if it is to be approved. Treatments must represent 'value for money' and a net benefit to society.
Orphan drugs
There are special rules for certain rare diseases ("orphan diseases") in several major drug regulatory territories. For example, diseases involving fewer than 200,000 patients in the United States, or larger populations in certain circumstances are subject to the Orphan Drug Act. Because medical research and development of drugs to treat such diseases is financially disadvantageous, companies that do so are rewarded with tax reductions, fee waivers, and market exclusivity on that drug for a limited time (seven years), regardless of whether the drug is protected by patents.
Global sales
| Company | Pharma revenue ($ million) |
|---|---|
|
Pfizer |
100,330 |
|
Johnson & Johnson |
94,940 |
|
Roche |
66,260 |
|
Merck & Co |
59,280 |
|
Abbvie |
58,050 |
|
Novartis |
50,540 |
|
Bristol Myers Squibb |
46,160 |
|
Sanofi |
45.220 |
|
AstraZeneca |
44,350 |
|
GSK |
36.150 |
|
Takeda |
30,000 |
|
Eli Lilly and Company |
28,550 |
|
Gilead Sciences |
27,280 |
|
Bayer |
26,640 |
|
Amgen |
26,320 |
|
Boehringer Ingelheim |
25,280 |
|
Novo Nordisk |
25,000 |
|
Moderna |
19,260 |
|
Merck KGaA |
19.160 |
|
BioNTech |
18,200 |
In 2011, global spending on prescription drugs topped $954 billion, even as growth slowed somewhat in Europe and North America. The United States accounts for more than a third of the global pharmaceutical market, with $340 billion in annual sales followed by the EU and Japan. Emerging markets such as China, Russia, South Korea and Mexico outpaced that market, growing a huge 81 percent.
The top ten best-selling drugs of 2013 totaled $75.6 billion in sales, with the anti-inflammatory drug Humira being the best-selling drug worldwide at $10.7 billion in sales. The second and third best selling were Enbrel and Remicade, respectively. The top three best-selling drugs in the United States in 2013 were Abilify ($6.3 billion,) Nexium ($6 billion) and Humira ($5.4 billion). The best-selling drug ever, Lipitor, averaged $13 billion annually and netted $141 billion total over its lifetime before Pfizer's patent expired in November 2011.
IMS Health publishes an analysis of trends expected in the pharmaceutical industry in 2007, including increasing profits in most sectors despite loss of some patents, and new 'blockbuster' drugs on the horizon.
Patents and generics
Depending on a number of considerations, a company may apply for and be granted a patent for the drug, or the process of producing the drug, granting exclusivity rights typically for about 20 years. However, only after rigorous study and testing, which takes 10 to 15 years on average, will governmental authorities grant permission for the company to market and sell the drug. Patent protection enables the owner of the patent to recover the costs of research and development through high profit margins for the branded drug. When the patent protection for the drug expires, a generic drug is usually developed and sold by a competing company. The development and approval of generics is less expensive, allowing them to be sold at a lower price. Often the owner of the branded drug will introduce a generic version before the patent expires in order to get a head start in the generic market. Restructuring has therefore become routine, driven by the patent expiration of products launched during the industry's "golden era" in the 1990s and companies' failure to develop sufficient new blockbuster products to replace lost revenues.
Prescriptions
In the U.S., the value of prescriptions increased over the period of 1995 to 2005 by 3.4 billion annually, a 61 percent increase. Retail sales of prescription drugs jumped 250 percent from $72 billion to $250 billion, while the average price of prescriptions more than doubled from $30 to $68.
Marketing
Advertising is common in healthcare journals as well as through more mainstream media routes. In some countries, notably the US, they are allowed to advertise directly to the general public. Pharmaceutical companies generally employ salespeople (often called 'drug reps' or, an older term, 'detail men') to market directly and personally to physicians and other healthcare providers. In some countries, notably the US, pharmaceutical companies also employ lobbyists to influence politicians. Marketing of prescription drugs in the US is regulated by the federal Prescription Drug Marketing Act of 1987. The pharmaceutical marketing plan incorporates the spending plans, channels, and thoughts which will take the drug association, and its items and administrations, forward in the current scene.
To healthcare professionals
The book Bad Pharma also discusses the influence of drug representatives, how ghostwriters are employed by the drug companies to write papers for academics to publish, how independent the academic journals really are, how the drug companies finance doctors' continuing education, and how patients' groups are often funded by industry.
Direct to consumer advertising
Since the 1980s, new methods of marketing for prescription drugs to consumers have become important. Direct-to-consumer media advertising was legalised in the FDA Guidance for Industry on Consumer-Directed Broadcast Advertisements.
Controversies
Drug marketing and lobbying
There has been increasing controversy surrounding pharmaceutical marketing and influence. There have been accusations and findings of influence on doctors and other health professionals through drug reps including the constant provision of marketing 'gifts' and biased information to health professionals; highly prevalent advertising in journals and conferences; funding independent healthcare organizations and health promotion campaigns; lobbying physicians and politicians (more than any other industry in the US); sponsorship of medical schools or nurse training; sponsorship of continuing educational events, with influence on the curriculum; and hiring physicians as paid consultants on medical advisory boards.
Some advocacy groups, such as No Free Lunch and AllTrials, have criticized the effect of drug marketing to physicians because they say it biases physicians to prescribe the marketed drugs even when others might be cheaper or better for the patient.
There have been related accusations of disease mongering (over-medicalizing) to expand the market for medications. An inaugural conference on that subject took place in Australia in 2006. In 2009, the Government-funded National Prescribing Service launched the "Finding Evidence – Recognizing Hype" program, aimed at educating GPs on methods for independent drug analysis.
Meta-analyses have shown that psychiatric studies sponsored by pharmaceutical companies are several times more likely to report positive results, and if a drug company employee is involved the effect is even larger. Influence has also extended to the training of doctors and nurses in medical schools, which is being fought.
It has been argued that the design of the Diagnostic and Statistical Manual of Mental Disorders and the expansion of the criteria represents an increasing medicalization of human nature, or "disease mongering", driven by drug company influence on psychiatry. The potential for direct conflict of interest has been raised, partly because roughly half the authors who selected and defined the DSM-IV psychiatric disorders had or previously had financial relationships with the pharmaceutical industry.
In the US, starting in 2013, under the Physician Financial Transparency Reports (part of the Sunshine Act), the Centers for Medicare & Medicaid Services has to collect information from applicable manufacturers and group purchasing organizations in order to report information about their financial relationships with physicians and hospitals. Data are made public in the Centers for Medicare & Medicaid Services website. The expectation is that relationship between doctors and Pharmaceutical industry will become fully transparent.
In a report conducted by OpenSecrets, there were more than 1,100 lobbyists working in some capacity for the pharmaceutical business in 2017. In the first quarter of 2017, the health products and pharmaceutical industry spent $78 million on lobbying members of the United States Congress.
Medication pricing
The pricing of pharmaceuticals is becoming a major challenge for health systems. A November 2020 study by the West Health Policy Center stated that more than 1.1 million senior citizens in the U.S. Medicare program are expected to die prematurely over the next decade because they will be unable to afford their prescription medications, requiring an additional $17.7 billion to be spent annually on avoidable medical costs due to health complications.
Regulatory issues
Ben Goldacre has argued that regulators – such as the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK, or the Food and Drug Administration (FDA) in the United States – advance the interests of the drug companies rather than the interests of the public due to revolving door exchange of employees between the regulator and the companies and friendships develop between regulator and company employees. He argues that regulators do not require that new drugs offer an improvement over what is already available, or even that they be particularly effective.
Others have argued that excessive regulation suppresses therapeutic innovation and that the current cost of regulator-required clinical trials prevents the full exploitation of new genetic and biological knowledge for the treatment of human disease. A 2012 report by the President's Council of Advisors on Science and Technology made several key recommendations to reduce regulatory burdens to new drug development, including 1) expanding the FDA's use of accelerated approval processes, 2) creating an expedited approval pathway for drugs intended for use in narrowly defined populations, and 3) undertaking pilot projects designed to evaluate the feasibility of a new, adaptive drug approval process.
Pharmaceutical fraud
Pharmaceutical fraud involves deceptions which bring financial gain to a pharmaceutical company. It affects individuals and public and private insurers. There are several different schemes used to defraud the health care system which are particular to the pharmaceutical industry. These include: Good Manufacturing Practice (GMP) Violations, Off Label Marketing, Best Price Fraud, CME Fraud, Medicaid Price Reporting, and Manufactured Compound Drugs. Of this amount $2.5 billion was recovered through False Claims Act cases in FY 2010. Examples of fraud cases include the GlaxoSmithKline $3 billion settlement, Pfizer $2.3 billion settlement and Merck & Co. $650 million settlement. Damages from fraud can be recovered by use of the False Claims Act, most commonly under the qui tam provisions which rewards an individual for being a "whistleblower", or relator (law).
Every major company selling atypical antipsychotics—Bristol-Myers Squibb, Eli Lilly and Company, Pfizer, AstraZeneca and Johnson & Johnson—has either settled recent government cases, under the False Claims Act, for hundreds of millions of dollars or is currently under investigation for possible health care fraud. Following charges of illegal marketing, two of the settlements set records in 2009 for the largest criminal fines ever imposed on corporations. One involved Eli Lilly's antipsychotic Zyprexa, and the other involved Bextra, an anti-inflammatory medication used for arthritis. In the Bextra case, the government also charged Pfizer with illegally marketing another antipsychotic, Geodon; Pfizer settled that part of the claim for $301 million, without admitting any wrongdoing.
On 2 July 2012, GlaxoSmithKline pleaded guilty to criminal charges and agreed to a $3 billion settlement of the largest health-care fraud case in the U.S. and the largest payment by a drug company. The settlement is related to the company's illegal promotion of prescription drugs, its failure to report safety data,bribing doctors, and promoting medicines for uses for which they were not licensed. The drugs involved were Paxil, Wellbutrin, Advair, Lamictal, and Zofran for off-label, non-covered uses. Those and the drugs Imitrex, Lotronex, Flovent, and Valtrex were involved in the kickback scheme.
The following is a list of the four largest settlements reached with pharmaceutical companies from 1991 to 2012, rank ordered by the size of the total settlement. Legal claims against the pharmaceutical industry have varied widely over the past two decades, including Medicare and Medicaid fraud, off-label promotion, and inadequate manufacturing practices.
| Company | Settlement | Violation(s) | Year | Product(s) | Laws allegedly violated (if applicable) |
|---|---|---|---|---|---|
| GlaxoSmithKline | $3 billion | Off-label promotion/ failure to disclose safety data |
2012 | Avandia/Wellbutrin/Paxil | False Claims Act/FDCA |
| Pfizer | $2.3 billion | Off-label promotion/kickbacks | 2009 |
Bextra/Geodon/ Zyvox/Lyrica |
False Claims Act/FDCA |
| Abbott Laboratories | $1.5 billion | Off-label promotion | 2012 | Depakote | False Claims Act/FDCA |
| Eli Lilly | $1.4 billion | Off-label promotion | 2009 | Zyprexa | False Claims Act/FDCA |
Physician roles
In May 2015, the New England Journal of Medicine emphasized the importance of pharmaceutical industry-physician interactions for the development of novel treatments, and argued that moral outrage over industry malfeasance had unjustifiably led many to overemphasize the problems created by financial conflicts of interest. The article noted that major healthcare organizations, such as National Center for Advancing Translational Sciences of the National Institutes of Health, the President's Council of Advisors on Science and Technology, the World Economic Forum, the Gates Foundation, the Wellcome Trust, and the Food and Drug Administration had encouraged greater interactions between physicians and industry in order to improve benefits to patients.
Response to COVID-19
In November 2020 several pharmaceutical companies announced successful trials of COVID-19 vaccines, with efficacy of 90 to 95% in preventing infection. Per company announcements and data reviewed by external analysts, these vaccines are priced at $3 to $37 per dose. The Wall Street Journal ran an editorial calling for this achievement to be recognized with a Nobel Peace Prize.
Doctors Without Borders warned that high prices and monopolies on medicines, tests, and vaccines would prolong the pandemic and cost lives. They urged governments to prevent profiteering, using compulsory licenses as needed, as had already been done by Canada, Chile, Ecuador, Germany, and Israel.
On 20 February, 46 US lawmakers called for the US government not to grant monopoly rights when giving out taxpayer development money for any coronavirus vaccines and treatments, to avoid giving exclusive control of prices and availability to private manufacturers.
In the United States the government signed agreements in which research and development and/or the building of manufacturing plants for potential COVID-19 therapeutics was subsidized. Typically, the agreement involved the government taking ownership of a certain number of doses of the product without further payment. For example, under the auspices of Operation Warp Speed in the United States, the government subsidized research related to COVID-19 vaccines and therapeutics at Regeneron, Johnson and Johnson, Moderna, AstraZeneca, Novavax, Pfizer, and GSK. Typical terms involved research subsidies of $400 million to $2 billion, and included government ownership of the first 100 million doses of any COVID-19 vaccine successfully developed.
American pharmaceutical company Gilead sought and obtained orphan drug status for remdesivir from the US Food and Drug Administration (FDA) on 23 March 2020. This provision is intended to encourage the development of drugs affecting fewer than 200,000 Americans by granting strengthened and extended legal monopoly rights to the manufacturer, along with waivers on taxes and government fees. Remdesivir is a candidate for treating COVID-19; at the time the status was granted, fewer than 200,000 Americans had COVID-19, but numbers were climbing rapidly as the COVID-19 pandemic reached the US, and crossing the threshold soon was considered inevitable. Remdesivir was developed by Gilead with over $79 million in U.S. government funding. In May 2020, Gilead announced that it would provide the first 940,000 doses of remdesivir to the federal government free of charge. After facing strong public reactions, Gilead gave up the "orphan drug" status for remdesivir on 25 March. Gilead retains 20-year remdesivir patents in more than 70 countries. In May 2020, the company further announced that it was in discussions with several generics companies to provide rights to produce remdesivir for developing countries, and with the Medicines Patent Pool to provide broader generic access.
Developing world
Patents
Patents have been criticized in the developing world, as they are thought to reduce access to existing medicines. Reconciling patents and universal access to medicine would require an efficient international policy of price discrimination. Moreover, under the TRIPS agreement of the World Trade Organization, countries must allow pharmaceutical products to be patented. In 2001, the WTO adopted the Doha Declaration, which indicates that the TRIPS agreement should be read with the goals of public health in mind, and allows some methods for circumventing pharmaceutical monopolies: via compulsory licensing or parallel imports, even before patent expiration.
In March 2001, 40 multi-national pharmaceutical companies brought litigation against South Africa for its Medicines Act, which allowed the generic production of antiretroviral drugs (ARVs) for treating HIV, despite the fact that these drugs were on-patent. HIV was and is an epidemic in South Africa, and ARVs at the time cost between US$10,000 and US$15,000 per patient per year. This was unaffordable for most South African citizens, and so the South African government committed to providing ARVs at prices closer to what people could afford. To do so, they would need to ignore the patents on drugs and produce generics within the country (using a compulsory license), or import them from abroad. After international protest in favour of public health rights (including the collection of 250,000 signatures by Médecins Sans Frontières), the governments of several developed countries (including The Netherlands, Germany, France, and later the US) backed the South African government, and the case was dropped in April of that year.
In 2016, GlaxoSmithKline (the world's sixth largest pharmaceutical company) announced that it would be dropping its patents in poor countries so as to allow independent companies to make and sell versions of its drugs in those areas, thereby widening the public access to them. GlaxoSmithKline published a list of 50 countries they would no longer hold patents in, affecting one billion people worldwide.
Charitable programs
In 2011 four of the top 20 corporate charitable donations and eight of the top 30 corporate charitable donations came from pharmaceutical manufacturers. The bulk of corporate charitable donations (69% as of 2012) comes by way of non-cash charitable donations, the majority of which again were donations contributed by pharmaceutical companies.
Charitable programs and drug discovery & development efforts by pharmaceutical companies include:
- "Merck's Gift", wherein billions of river blindness drugs were donated in Africa
- Pfizer's gift of free/discounted fluconazole and other drugs for AIDS in South Africa
- GSK's commitment to give free albendazole tablets to the WHO for, and until, the elimination of lymphatic filariasis worldwide.
- In 2006, Novartis committed US$755 million in corporate citizenship initiatives around the world, particularly focusing on improving access to medicines in the developing world through its Access to Medicine projects, including donations of medicines to patients affected by leprosy, tuberculosis, and malaria; Glivec patient assistance programs; and relief to support major humanitarian organisations with emergency medical needs.
See also
- List of industrial complexes – Economic concept
- Big Pharma conspiracy theory – Conspiracy theories about the pharmaceutical industry
- Clinical trial – Phase of clinical research in medicine
- Drug development – Process of bringing a new pharmaceutical drug to the market
- Drug discovery – Pharmaceutical procedure
- Legal drug trade – manufacture and sale of pharmaceutical drugs
- List of pharmaceutical companies
- Licensed production – Production under license of technology developed elsewhere
- Outsourcing – Contracting formerly internal tasks to an external organization
- Pharmaceutical marketing – Advertising by pharmaceutical companies
- Pharmacy – Clinical health science
- Pharmacy benefit management – Administration of prescription drug programs in the United States
- Unitaid – Global health initiative
- Valuation (finance) § Valuation of intangible assets
External links
-
 Quotations related to Pharmaceutical industry at Wikiquote
Quotations related to Pharmaceutical industry at Wikiquote - "Global Medicines Use in 2020". IMS Institute for Healthcare Informatics. November 2015.
- "The pharmaceutical industry and global health – Facts & figures 2017" (PDF). International Federation of Pharmaceutical Manufacturers & Associations. February 2017.
- "Licensing Agreements in the Pharmaceutical Industry".
- "The Pharmaceutical Industry in Figures – Key Data 2018" (PDF). European Federation of Pharmaceutical Industries and Associations.
|
Pharmaceutical industry in the United States
| |
|---|---|
| Companies | |
| Regulatory bodies | |
| Professional bodies | |
| Miscellaneous | |
| Books |
|
| Lists | |
| Related templates | |
| Related categories |
|
| General | |
|---|---|
| Pharmaceutical sciences | |
| Professions | |
| Practice areas | |
| Pharmaceutical industry | |
|
Major industries
| |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
| |||||||||||||||||||||
| |||||||||||||||||||||
| |||||||||||||||||||||
| |||||||||||||||||||||
| General |
|
||||||
|---|---|---|---|---|---|---|---|
| Preventive healthcare | |||||||
| Population health | |||||||
| Biological and epidemiological statistics |
|||||||
| Infectious and epidemic disease prevention |
|||||||
| Food hygiene and safety management |
|||||||
| Health behavioral sciences |
|||||||
| Organizations, education and history |
|
||||||
|
Specialties and subspecialties |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Medical education |
|||||||||||
| Related topics | |||||||||||