
Phylogenetic comparative methods (PCMs) use information on the historical relationships of lineages (phylogenies) to test evolutionary hypotheses. The comparative method has a long history in evolutionary biology; indeed, Charles Darwin used differences and similarities between species as a major source of evidence in The Origin of Species. However, the fact that closely related lineages share many traits and trait combinations as a result of the process of descent with modification means that lineages are not independent. This realization inspired the development of explicitly phylogenetic comparative methods. Initially, these methods were primarily developed to control for phylogenetic history when testing for adaptation; however, in recent years the use of the term has broadened to include any use of phylogenies in statistical tests. Although most studies that employ PCMs focus on extant organisms, many methods can also be applied to extinct taxa and can incorporate information from the fossil record.
PCMs can generally be divided into two types of approaches: those that infer the evolutionary history of some character (phenotypic or genetic) across a phylogeny and those that infer the process of evolutionary branching itself (diversification rates), though there are some approaches that do both simultaneously. Typically the tree that is used in conjunction with PCMs has been estimated independently (see computational phylogenetics) such that both the relationships between lineages and the length of branches separating them is assumed to be known.
Applications
Phylogenetic comparative approaches can complement other ways of studying adaptation, such as studying natural populations, experimental studies, and mathematical models. Interspecific comparisons allow researchers to assess the generality of evolutionary phenomena by considering independent evolutionary events. Such an approach is particularly useful when there is little or no variation within species. And because they can be used to explicitly model evolutionary processes occurring over very long time periods, they can provide insight into macroevolutionary questions, once the exclusive domain of paleontology.


Phylogenetic comparative methods are commonly applied to such questions as:
- What is the slope of an allometric scaling relationship?
→ Example: how does brain mass vary in relation to body mass?
- Do different clades of organisms differ with respect to some phenotypic trait?
→ Example: do canids have larger hearts than felids?
- Do groups of species that share a behavioral or ecological feature (e.g., social system, diet) differ in average phenotype?
→ Example: do carnivores have larger home ranges than herbivores?
→ Example: where did endothermy evolve in the lineage that led to mammals?
→ Example: where, when, and why did placentas and viviparity evolve?
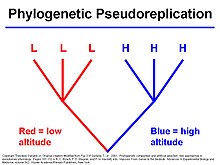
- Does a trait exhibit significant phylogenetic signal in a particular group of organisms? Do certain types of traits tend to "follow phylogeny" more than others?
→ Example: are behavioral traits more labile during evolution?
- Do species differences in life history traits trade-off, as in the so-called fast-slow continuum?
→ Example: why do small-bodied species have shorter life spans than their larger relatives?
Phylogenetically independent contrasts

Felsenstein proposed the first general statistical method in 1985 for incorporating phylogenetic information, i.e., the first that could use any arbitrary topology (branching order) and a specified set of branch lengths. The method is now recognized as an algorithm that implements a special case of what are termed phylogenetic generalized least-squares models. The logic of the method is to use phylogenetic information (and an assumed Brownian motion like model of trait evolution) to transform the original tip data (mean values for a set of species) into values that are statistically independent and identically distributed.
The algorithm involves computing values at internal nodes as an intermediate step, but they are generally not used for inferences by themselves. An exception occurs for the basal (root) node, which can be interpreted as an estimate of the ancestral value for the entire tree (assuming that no directional evolutionary trends [e.g., Cope's rule] have occurred) or as a phylogenetically weighted estimate of the mean for the entire set of tip species (terminal taxa). The value at the root is equivalent to that obtained from the "squared-change parsimony" algorithm and is also the maximum likelihood estimate under Brownian motion. The independent contrasts algebra can also be used to compute a standard error or confidence interval.
Phylogenetic generalized least squares (PGLS)
Probably the most commonly used PCM is phylogenetic generalized least squares (PGLS). This approach is used to test whether there is a relationship between two (or more) variables while accounting for the fact that lineage are not independent. The method is a special case of generalized least squares (GLS) and as such the PGLS estimator is also unbiased, consistent, efficient, and asymptotically normal. In many statistical situations where GLS (or, ordinary least squares [OLS]) is used residual errors ε are assumed to be independent and identically distributed random variables that are assumed to be normal

whereas in PGLS the errors are assumed to be distributed as

where V is a matrix of expected variance and covariance of the residuals given an evolutionary model and a phylogenetic tree. Therefore, it is the structure of residuals and not the variables themselves that show phylogenetic signal. This has long been a source of confusion in the scientific literature. A number of models have been proposed for the structure of V such as Brownian motionOrnstein-Uhlenbeck, and Pagel's λ model. (When a Brownian motion model is used, PGLS is identical to the independent contrasts estimator.). In PGLS, the parameters of the evolutionary model are typically co-estimated with the regression parameters.
PGLS can only be applied to questions where the dependent variable is continuously distributed; however, the phylogenetic tree can also be incorporated into the residual distribution of generalized linear models, making it possible to generalize the approach to a broader set of distributions for the response.
Phylogenetically informed Monte Carlo computer simulations

Martins and Garland proposed in 1991 that one way to account for phylogenetic relations when conducting statistical analyses was to use computer simulations to create many data sets that are consistent with the null hypothesis under test (e.g., no correlation between two traits, no difference between two ecologically defined groups of species) but that mimic evolution along the relevant phylogenetic tree. If such data sets (typically 1,000 or more) are analyzed with the same statistical procedure that is used to analyze a real data set, then results for the simulated data sets can be used to create phylogenetically correct (or "PC") null distributions of the test statistic (e.g., a correlation coefficient, t, F). Such simulation approaches can also be combined with such methods as phylogenetically independent contrasts or PGLS (see above).
See also
- Allometry
- Behavioral ecology
- Biodiversity
- Bioinformatics
- Cladistics
- Comparative anatomy
- Comparative method in linguistics
- Comparative physiology
- Computational phylogenetics
- Disk-covering method
- Ecophysiology
- Evolutionary neurobiology
- Evolutionary physiology
- Generalized least squares (GLS)
- Generalized linear model
- Joe Felsenstein
- Mark Pagel
- Maximum likelihood
- Maximum parsimony
- Paul H. Harvey
- Phylogenetics
- Phylogenetic reconciliation
- Roderic D.M. Page
- Sexual selection
- Statistics
- Systematics
- Theodore Garland Jr.
Further reading
- Ackerly, D. D. 1999. Comparative plant ecology and the role of phylogenetic information. Pages 391–413 in M. C. Press, J. D. Scholes, and M. G. Braker, eds. Physiological plant ecology. The 39th symposium of the British Ecological Society held at the University of York 7–9 September 1998. Blackwell Science, Oxford, U.K.
- Berenbrink, M.; Koldkjær, P.; Kepp, O.; Cossins, A. R. (2005). "Evolution of oxygen secretion in fishes and the emergence of a complex physiological system". Science. 307 (5716): 1752–1757. Bibcode:2005Sci...307.1752B. doi:10.1126/science.1107793. PMID 15774753. S2CID 36391252.
- Blomberg, S. P.; Garland, T. Jr.; Ives, A. R. (2003). "Testing for phylogenetic signal in comparative data: behavioral traits are more labile" (PDF). Evolution. 57 (4): 717–745. doi:10.1554/0014-3820(2003)057[0717:tfpsic]2.0.co;2. PMID 12778543.
- Brooks, D. R., and D. A. McLennan. 1991. Phylogeny, ecology, and behavior: a research program in comparative biology. Univ. Chicago Press, Chicago. 434 pp.
- Cheverud, J. M.; Dow, M. M.; Leutenegger, W. (1985). "The quantitative assessment of phylogenetic constraints in comparative analyses: sexual dimorphism in body weight among primates". Evolution. 39 (6): 1335–1351. doi:10.2307/2408790. JSTOR 2408790. PMID 28564267.
- Eggleton, P., and R. I. Vane-Wright, eds. 1994. Phylogenetics and ecology. Linnean Society Symposium Series Number 17. Academic Press, London.
- Felsenstein, J. 2004. Inferring phylogenies. Sinauer Associates, Sunderland, Mass. xx + 664 pp.
- Freckleton, R. P.; Harvey, P. H.; Pagel, M. (2002). "Phylogenetic analysis and comparative data: a test and review of evidence". American Naturalist. 160 (6): 712–726. doi:10.1086/343873. PMID 18707460. S2CID 19796539.
- Garland, T. Jr.; Ives, A. R. (2000). "Using the past to predict the present: Confidence intervals for regression equations in phylogenetic comparative methods" (PDF). American Naturalist. 155 (3): 346–364. doi:10.1086/303327. PMID 10718731. S2CID 4384701.
- Garland, T. Jr.; Bennett, A. F.; Rezende, E. L. (2005). "Phylogenetic approaches in comparative physiology" (PDF). Journal of Experimental Biology. 208 (16): 3015–3035. doi:10.1242/jeb.01745. PMID 16081601. S2CID 14871059.
- Garland, T. Jr.; Harvey, P. H.; Ives, A. R. (1992). "Procedures for the analysis of comparative data using phylogenetically independent contrasts" (PDF). Systematic Biology. 41 (1): 18–32. doi:10.2307/2992503. JSTOR 2992503.
- Gittleman, J. L.; Kot, M. (1990). "Adaptation: statistics and a null model for estimating phylogenetic effects". Systematic Zoology. 39 (3): 227–241. doi:10.2307/2992183. JSTOR 2992183.
- Hadfield, J. D; Nakagawa, S. (2010). "General quantitative genetic methods for comparative biology: phylogenies, taxonomies and multi-trait models for continuous and categorical characters". Journal of Evolutionary Biology. 23 (3): 494–508. doi:10.1111/j.1420-9101.2009.01915.x. PMID 20070460. S2CID 27706318.
-
Herrada, E. A.; Tessone, C. J.; Klemm, K.; Eguiluz, V. M.; Hernandez-Garcia, E.; Duarte, C. M. (2008). "Universal Scaling in the Branching of the Tree of Life". PLOS ONE. 3 (7): e2757. arXiv:0807.4042. Bibcode:2008PLoSO...3.2757H. doi:10.1371/journal.pone.0002757. PMC 2447175. PMID 18648500.

- Housworth, E. A.; Martins, E. P.; Lynch, M. (2004). "The phylogenetic mixed model" (PDF). American Naturalist. 163 (1): 84–96. doi:10.1086/380570. PMID 14767838. S2CID 10568814.
- Ives, A. R. 2018. Mixed and phylogenetic models: a conceptual introduction to correlated data. leanpub.com, 125 pp., https://leanpub.com/correlateddata
- Ives, A. R.; Midford, P. E.; Garland, T. Jr. (2007). "Within-species variation and measurement error in phylogenetic comparative methods". Systematic Biology. 56 (2): 252–270. doi:10.1080/10635150701313830. PMID 17464881.
- Maddison, D. R. (1994). "Phylogenetic methods for inferring the evolutionary history and process of change in discretely valued characters". Annual Review of Entomology. 39: 267–292. doi:10.1146/annurev.ento.39.1.267.
- Maddison, W. P. (1990). "A method for testing the correlated evolution of two binary characters: Are gains or losses concentrated on certain branches of a phylogenetic tree?". Evolution. 44 (3): 539–557. doi:10.2307/2409434. JSTOR 2409434. PMID 28567979.
- Maddison, W. P., and D. R. Maddison. 1992. MacClade. Analysis of phylogeny and character evolution. Version 3. Sinauer Associates, Sunderland, Mass. 398 pp.
- Martins, E. P., ed. 1996. Phylogenies and the comparative method in animal behavior. Oxford University Press, Oxford. 415 pp.
- Martins, E. P.; Hansen, T. F. (1997). "Phylogenies and the comparative method: a general approach to incorporating phylogenetic information into the analysis of interspecific data". American Naturalist. 149 (4): 646–667. doi:10.1086/286013. S2CID 29362369. Erratum Am. Nat. 153:448.
- Nunn, C. L.; Barton, R. A. (2001). "Comparative methods for studying primate adaptation and allometry". Evolutionary Anthropology. 10 (3): 81–98. doi:10.1002/evan.1019. S2CID 16959643.
- Oakley, T. H.; Gu, Z.; Abouheif, E.; Patel, N. H.; Li, W.-H. (2005). "Comparative methods for the analysis of gene-expression evolution: an example using yeast functional genomic data" (PDF). Molecular Biology and Evolution. 22: 40–50. doi:10.1093/molbev/msh257. PMID 15356281.
- O'Meara, B. C.; Ané, C. M.; Sanderson, M. J.; Wainwright, P. C. (2006). "Testing for different rates of continuous trait evolution in different groups using likelihood" (PDF). Evolution. 60: 922–933. doi:10.1554/05-130.1. S2CID 13796463.
- Organ, C. L.; Shedlock, A. M.; Meade, A.; Pagel, M.; Edwards, S. V. (2007). "Origin of avian genome size and structure in non-avian dinosaurs". Nature. 446 (7132): 180–184. Bibcode:2007Natur.446..180O. doi:10.1038/nature05621. PMID 17344851. S2CID 3031794.
- Page, R. D. M., ed. 2003. Tangled trees: phylogeny, cospeciation, and coevolution. University of Chicago Press, Chicago.
- Pagel, M. D. (1993). "Seeking the evolutionary regression coefficient: an analysis of what comparative methods measure". Journal of Theoretical Biology. 164 (2): 191–205. doi:10.1006/jtbi.1993.1148. PMID 8246516.
- Pagel, M (1999). "Inferring the historical patterns of biological evolution". Nature. 401 (6756): 877–884. Bibcode:1999Natur.401..877P. doi:10.1038/44766. hdl:2027.42/148253. PMID 10553904. S2CID 205034365.
- Paradis, E (2005). "Statistical analysis of diversification with species traits" (PDF). Evolution. 59 (1): 1–12. doi:10.1554/04-231. PMID 15792222. S2CID 196612333.
- Paradis, E.; Claude, J. (2002). "Analysis of comparative data using generalized estimating equations" (PDF). Journal of Theoretical Biology. 218 (2): 175–185. doi:10.1006/jtbi.2002.3066. PMID 12381290.
- Purvis, A.; Garland, T. Jr. (1993). "Polytomies in comparative analyses of continuous characters" (PDF). Systematic Biology. 42 (4): 569–575. doi:10.2307/2992489. JSTOR 2992489.
- Rezende, E. L., and Garland, T. Jr. 2003. Comparaciones interespecíficas y métodos estadísticos filogenéticos. Pages 79–98 in F. Bozinovic, ed. Fisiología Ecológica & Evolutiva. Teoría y casos de estudios en animales. Ediciones Universidad Católica de Chile, Santiago. PDF
- Rezende, E.L.; Diniz-Filho, J.A.F (2012). "Phylogenetic analyses: comparing species to infer adaptations and physiological mechanisms" (PDF). Comprehensive Physiology. 2 (1): 639–674. doi:10.1002/cphy.c100079. PMID 23728983.
- Ridley, M. 1983. The explanation of organic diversity: The comparative method and adaptations for mating. Clarendon, Oxford, U.K.
- Rohlf, F. J. (2001). "Comparative methods for the analysis of continuous variables: geometric interpretations". Evolution. 55 (11): 2143–2160. doi:10.1111/j.0014-3820.2001.tb00731.x. PMID 11794776. S2CID 23200090.
- Rohlf, F. J. (2006). "A comment on phylogenetic correction". Evolution. 60 (7): 1509–1515. doi:10.1554/05-550.1. PMID 16929667. S2CID 198156300.
- Sanford, G. M.; Lutterschmidt, W. I.; Hutchison, V. H. (2002). "The comparative method revisited". BioScience. 52 (9): 830–836. doi:10.1641/0006-3568(2002)052[0830:tcmr]2.0.co;2.
- Schluter, D.; Price, T.; Mooers, A. O.; Ludwig, D. (1997). "Likelihood of ancestor states in adaptive radiation". Evolution. 51 (6): 1699–1711. doi:10.2307/2410994. JSTOR 2410994. PMID 28565128.
- Smith, R. J.; Cheverud, J. M. (2002). "Scaling of sexual size dimorphism in body mass: a phylogenetic analysis of Rensch's rule in primates". International Journal of Primatology. 23 (5): 1095–1135. doi:10.1023/A:1019654100876. S2CID 42439809.
- Steppan, S. J.; Phillips, P. C.; Houle, D. (2002). "Comparative quantitative genetics: evolution of the G matrix" (PDF). Trends in Ecology and Evolution. 17 (7): 320–327. doi:10.1016/s0169-5347(02)02505-3.
- Vanhooydonck, B.; Van Damme, R. (1999). "Evolutionary relationships between body shape and habitat use in lacertid lizards". Evolutionary Ecology Research. 1: 785–805.
External links
- Adaptation and the comparative method online lecture, with worked example of phylogenetically independent contrasts and mastery quiz
- List of phylogeny programs
- Phylocomm
- Phylogenetic Tools for Comparative Biology
- Phylogeny of Sleep website
- Tree of Life
Journals
- American Naturalist
- Behavioral Ecology
- Ecology
- Evolution
- Evolutionary Ecology Research
- Functional Ecology
- Journal of Evolutionary Biology
- Philosophical Transactions of the Royal Society of London B
- Physiological and Biochemical Zoology
- Systematic Biology
Software packages (incomplete list)
- Analyses of Phylogenetics and Evolution
- BayesTraits
- Comparative Analysis by Independent Contrasts
- COMPARE
- Felsenstein's List
- Mesquite PDAP:PDTree for Mesquite
- mvMorph
- ouch: Ornstein-Uhlenbeck for Comparative Hypotheses
- PDAP: Phenotypic Diversity Analysis Programs
- Phylocom
- Phylogenetic Regression
- PHYSIG
Laboratories
| Relevant fields | ||
|---|---|---|
| Basic concepts | ||
| Inference methods | ||
| Current topics | ||
| Group traits | ||
| Group types | ||
| Nomenclature | ||