
| Properdin deficiency | |
|---|---|
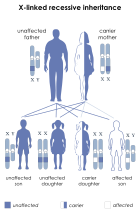 | |
| This condition is inherited in an x-linked recessive manner |
Properdin deficiency is a rare X-linked disease in which properdin, an important complement factor responsible for the stabilization of the alternative C3 convertase, is deficient. There are three forms of properdin deficiencies: Type I, which is identified by the total absence of the properdin protein in the plasma, Type II, which is a low but detectable amount of the properdin protein in the plasma, and Type III, which is a rare case of normal levels of properdin protein, but a dysfunctional variant. One of the first studied cases of properdin deficiency was in 1980 by Davis and Forrestal. These families had members with only partial deficiencies which resulted in a lowered consumption of the C3 protein. Properdin deficiency was studied again shortly after in 1982 by Sjoholm in which all of the subjects were deceased shortly after the study because of their disease. The largest study of properdin deficiency was in 1989 by Fijen which included nine males across three generations. Out of the 46 family members in Fijen's study, the 9 who were affected were found to be more susceptible to diseases from the Neisseria genus.
Signs and symptoms
As a protein involved in the function of the immune system, no external changes in physiology or aberrant physical characteristics are expressed by individuals possessing a properdin deficiency. However, individuals that have a properdin deficiency do have a heightened susceptibility to bacterial infections, most notably caused by bacteria within the Neisseria genus, though there have also been studied cases of individuals with recurrent pneumococcus bacteremia as a result of Streptococcus pneumoniae, another species of bacteria from an entirely different phylum. Due to a heightened susceptibility to Neisseria bacterium, individuals with properdin deficiency are far more likely to succumb to bacterial infection such as meningitis, resulting in inflammation of the brain and spinal cord, which causes severe headaches, fevers, and neck stiffness, and may result in further development of other meningococcal diseases and extreme complications such as sepsis. Individuals with properdin deficiency are also more likely to catch the sexually transmitted disease, gonorrhea, as it is also caused by Neisseria bacterium, resulting in swelling, itching, pain, and formation of pus on the mucous membranes, including, but not limited to, the genitals, mouth, and rectum.
Diagnosis
As mentioned before, there are no external indications of properdin deficiency, and as such, properdin deficiency can only be reliably detected by lab tests. The typical tests for complement deficiencies, such as the measurement of C3 and C4, do not detect low levels of the absence of properdin. These pathways are typically unaltered by any of the three types of properdin deficiencies, but even when they are affected, it is typically within normal levels and is not cause for concern. Instead, histories of infection with anything from the Neisseria genus as well as family history can be indicators, but only specialist centers can screen for properdin deficiencies using immunochemical assays. In particular, the use of ELISA proves to be one of the most effective methods of detecting properdin deficiency, as the average healthy male is expected to show properdin antigen levels of around 128.0 ELISA units/ml, and obligate carrier females (recall that properdin deficiency is an X-linked disease) tend to show an average of 45.6 units/ml. An individual with properdin deficiency should, by definition, show very little to no properdin antigen levels at all, as they do not possess the requisite gene to produce the protein. While properdin deficiencies are rare, they have only been diagnosed in Caucasians, but no other race/ethnicity.
Management
Pertaining to complement deficiencies, there is no cure and the treatments for complement deficiencies vary widely. The best course of action for management is usually for a patient to treat the complement deficiency as an immune deficiency and get immunized against the microbe associated with their deficiency (or best candidate). As mentioned earlier, individuals with properdin deficiency are increasingly susceptible to Neisseria bacterium. Recent studies have indicated that individuals with properdin deficiency respond well when they are immunized with tetravalent polysaccharide meningococcal vaccine, which generates anti capsular antibodies and bactericidal anti-meningococcal activity against serotypes covered by the given vaccine. The vaccine has been reported to lower the chances of reinfection by meningococci in individuals who have undergone the treatment, however the vaccine does not protect against group B meningococci and chemotherapy is recommended if full protection from all meningococci variants is desired.
Genetics
Properdin deficiency is caused by an X-linked recessive allele, meaning that the gene responsible is only present on the X chromosome. Given that it is recessive, this means that the condition can only be inherited if the alleles for both X-chromosomes have the deficiency if the patient is female (xx) or if patient only has one X chromosome, as with male patients (xY). This means that a female could be heterozygous for the allele (Xx) and not express the weakened immune system, as individuals with partial properdin deficiency have been shown to function effectively the same as healthy individuals. This also means that any male patient that receives the recessive allele from their mother will inherit the deficiency, regardless of the genotype of their father, as the father must have passed the Y chromosome to them, which plays no part in genes with x-linked inheritance.
The gene responsible for the production of properdin, Complement Factor Properdin (CFP), lies on the X-chromosome at the coordinates: GRCh38: X:47,623,281-47,630,304. There are three types of properdin deficiency, which are caused by different mutations in the exons of the CFP gene.
Type I properdin deficiency can be the result of a nonsense mutation on exon 5 of the CDP gene, caused by a C-to-T transition at position 2767, which results in an early termination of the sequence and the production of an immature properdin protein. Type I can also be the result of a G-T transversion at position 3511 in exon 7, causing an amino acid substitution from gly271 to valine, which renders the protein non-functional. Finally, Type I can alternatively be caused by a C-to-G transversion on exon 6 at position 3041, converting a serine codon to a stop codon.
Type II properdin deficiency is thought to be caused by one or both of two mutations on the CFP gene. One of the mutations is a C-to-T transition at position 2124, located on exon 4, which converted an arginine to a tryptophan, resulting in an improper protein fold. The second mutation is a G-to-A transition at position 827 in intron 3, which is hypothesized to cause a cryptic splice site, resulting in improper splicing of the mRNA.
Type III properdin deficiency is caused by a T-to-G mutation on exon 9, resulting in a conversion of tyr387 to aspartic acid, resulting in an ineffective protein.
Type I and type II properdin deficiency result in an absence or extremely low presence of properdin, whereas type III results in the presence of a dysfunctional properdin protein structure. Ultimately, all three types have the same basic effect, reducing defensive capabilities against bacteria, especially those previously mentioned.
Epidemiology
Complement deficiencies are rare and currently not well characterized, so there has been difficulty detecting them. Currently, complement deficiencies only comprise approximately 2% of all primary immunodeficiency disorders. While the frequency of properdin deficiency has not been assessed worldwide, the risk of meningococcal infection in an individual with properdin deficiency has been calculated to be around 50%.