

Carcinogenesis, also called oncogenesis or tumorigenesis, is the formation of a cancer, whereby normal cells are transformed into cancer cells. The process is characterized by changes at the cellular, genetic, and epigenetic levels and abnormal cell division. Cell division is a physiological process that occurs in almost all tissues and under a variety of circumstances. Normally, the balance between proliferation and programmed cell death, in the form of apoptosis, is maintained to ensure the integrity of tissues and organs. According to the prevailing accepted theory of carcinogenesis, the somatic mutation theory, mutations in DNA and epimutations that lead to cancer disrupt these orderly processes by interfering with the programming regulating the processes, upsetting the normal balance between proliferation and cell death. This results in uncontrolled cell division and the evolution of those cells by natural selection in the body. Only certain mutations lead to cancer whereas the majority of mutations do not.
Variants of inherited genes may predispose individuals to cancer. In addition, environmental factors such as carcinogens and radiation cause mutations that may contribute to the development of cancer. Finally random mistakes in normal DNA replication may result in cancer causing mutations. A series of several mutations to certain classes of genes is usually required before a normal cell will transform into a cancer cell. Recent comprehensive patient-level classification and quantification of driver events in TCGA cohorts revealed that there are on average 12 driver events per tumor, of which 0.6 are point mutations in oncogenes, 1.5 are amplifications of oncogenes, 1.2 are point mutations in tumor suppressors, 2.1 are deletions of tumor suppressors, 1.5 are driver chromosome losses, 1 is a driver chromosome gain, 2 are driver chromosome arm losses, and 1.5 are driver chromosome arm gains. Mutations in genes that regulate cell division, apoptosis (cell death), and DNA repair may result in uncontrolled cell proliferation and cancer.
Cancer is fundamentally a disease of regulation of tissue growth. In order for a normal cell to transform into a cancer cell, genes that regulate cell growth and differentiation must be altered. Genetic and epigenetic changes can occur at many levels, from gain or loss of entire chromosomes, to a mutation affecting a single DNA nucleotide, or to silencing or activating a microRNA that controls expression of 100 to 500 genes. There are two broad categories of genes that are affected by these changes. Oncogenes may be normal genes that are expressed at inappropriately high levels, or altered genes that have novel properties. In either case, expression of these genes promotes the malignant phenotype of cancer cells. Tumor suppressor genes are genes that inhibit cell division, survival, or other properties of cancer cells. Tumor suppressor genes are often disabled by cancer-promoting genetic changes. Finally Oncovirinae, viruses that contain an oncogene, are categorized as oncogenic because they trigger the growth of tumorous tissues in the host. This process is also referred to as viral transformation.
Causes
Genetic and epigenetic
There is a diverse classification scheme for the various genomic changes that may contribute to the generation of cancer cells. Many of these changes are mutations, or changes in the nucleotide sequence of genomic DNA. There are also many epigenetic changes that alter whether genes are expressed or not expressed. Aneuploidy, the presence of an abnormal number of chromosomes, is one genomic change that is not a mutation, and may involve either gain or loss of one or more chromosomes through errors in mitosis. Large-scale mutations involve either the deletion or duplication of a portion of a chromosome. Genomic amplification occurs when a cell gains many copies (often 20 or more) of a small chromosomal region, usually containing one or more oncogenes and adjacent genetic material. Translocation occurs when two separate chromosomal regions become abnormally fused, often at a characteristic location. A well-known example of this is the Philadelphia chromosome, or translocation of chromosomes 9 and 22, which occurs in chronic myelogenous leukemia, and results in production of the BCR-abl fusion protein, an oncogenic tyrosine kinase. Small-scale mutations include point mutations, deletions, and insertions, which may occur in the promoter of a gene and affect its expression, or may occur in the gene's coding sequence and alter the function or stability of its protein product. Disruption of a single gene may also result from integration of genomic material from a DNA virus or retrovirus, and such an event may also result in the expression of viral oncogenes in the affected cell and its descendants.
DNA damage

DNA damage is considered to be the primary cause of cancer. More than 60,000 new naturally-occurring instances of DNA damage arise, on average, per human cell, per day, due to endogenous cellular processes (see article DNA damage (naturally occurring)).
Additional DNA damage can arise from exposure to exogenous agents. As one example of an exogenous carcinogenic agent, tobacco smoke causes increased DNA damage, and this DNA damage likely cause the increase of lung cancer due to smoking. In other examples, UV light from solar radiation causes DNA damage that is important in melanoma,Helicobacter pylori infection produces high levels of reactive oxygen species that damage DNA and contribute to gastric cancer, and the Aspergillus flavus metabolite aflatoxin is a DNA damaging agent that is causative in liver cancer.
DNA damage can also be caused by substances produced in the body. Macrophages and neutrophils in an inflamed colonic epithelium are the source of reactive oxygen species causing the DNA damage that initiates colonic tumorigenesis, and bile acids, at high levels in the colons of humans eating a high-fat diet, also cause DNA damage and contribute to colon cancer.
Such exogenous and endogenous sources of DNA damage are indicated in the boxes at the top of the figure in this section. The central role of DNA damage in progression to cancer is indicated at the second level of the figure. The central elements of DNA damage, epigenetic alterations and deficient DNA repair in progression to cancer are shown in red.
A deficiency in DNA repair would cause more DNA damage to accumulate, and increase the risk for cancer. For example, individuals with an inherited impairment in any of 34 DNA repair genes (see article DNA repair-deficiency disorder) are at increased risk of cancer, with some defects causing an up to 100% lifetime chance of cancer (e.g. p53 mutations). Such germline mutations are shown in a box at the left of the figure, with an indication of their contribution to DNA repair deficiency. However, such germline mutations (which cause highly penetrant cancer syndromes) are the cause of only about one percent of cancers.
The majority of cancers are called non-hereditary or "sporadic cancers". About 30% of sporadic cancers do have some hereditary component that is currently undefined, while the majority, or 70% of sporadic cancers, have no hereditary component.
In sporadic cancers, a deficiency in DNA repair is occasionally due to a mutation in a DNA repair gene; much more frequently, reduced or absent expression of DNA repair genes is due to epigenetic alterations that reduce or silence gene expression. This is indicated in the figure at the 3rd level from the top. For example, for 113 colorectal cancers examined in sequence, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region (an epigenetic alteration).
When expression of DNA repair genes is reduced, this causes a DNA repair deficiency. This is shown in the figure at the 4th level from the top. With a DNA repair deficiency, DNA damage persists in cells at a higher than typical level (5th level from the top in figure); this excess damage causes an increased frequency of mutation and/or epimutation (6th level from top of figure). Experimentally, mutation rates increase substantially in cells defective in DNA mismatch repair or in Homologous recombinational repair (HRR).Chromosomal rearrangements and aneuploidy also increase in HRR-defective cells During repair of DNA double-strand breaks, or repair of other DNA damage, incompletely-cleared repair sites can cause epigenetic gene silencing.
The somatic mutations and epigenetic alterations caused by DNA damage and deficiencies in DNA repair accumulate in field defects. Field defects are normal-appearing tissues with multiple alterations (discussed in the section below), and are common precursors to development of the disordered and over-proliferating clone of tissue in a cancer. Such field defects (second level from bottom of figure) may have numerous mutations and epigenetic alterations.
It is impossible to determine the initial cause for most specific cancers. In a few cases, only one cause exists: for example, the virus HHV-8 causes all Kaposi's sarcomas. However, with the help of cancer epidemiology techniques and information, it is possible to produce an estimate of a likely cause in many more situations. For example, lung cancer has several causes, including tobacco use and radon gas. Men who currently smoke tobacco develop lung cancer at a rate 14 times that of men who have never smoked tobacco: the chance of lung cancer in a current smoker being caused by smoking is about 93%; there is a 7% chance that the smoker's lung cancer was caused by radon gas or some other, non-tobacco cause. These statistical correlations have made it possible for researchers to infer that certain substances or behaviors are carcinogenic. Tobacco smoke causes increased exogenous DNA damage, and this DNA damage is the likely cause of lung cancer due to smoking. Among the more than 5,000 compounds in tobacco smoke, the genotoxic DNA-damaging agents that occur both at the highest concentrations, and which have the strongest mutagenic effects are acrolein, formaldehyde, acrylonitrile, 1,3-butadiene, acetaldehyde, ethylene oxide and isoprene.
Using molecular biological techniques, it is possible to characterize the mutations, epimutations or chromosomal aberrations within a tumor, and rapid progress is being made in the field of predicting certain cancer patients' prognosis based on the spectrum of mutations. For example, up to half of all tumors have a defective p53 gene. This mutation is associated with poor prognosis, since those tumor cells are less likely to go into apoptosis or programmed cell death when damaged by therapy. Telomerase mutations remove additional barriers, extending the number of times a cell can divide. Other mutations enable the tumor to grow new blood vessels to provide more nutrients, or to metastasize, spreading to other parts of the body. However, once a cancer is formed it continues to evolve and to produce sub-clones. It was reported in 2012 that a single renal cancer specimen, sampled in nine different areas, had 40 "ubiquitous" mutations, found in all nine areas, 59 mutations shared by some, but not all nine areas, and 29 "private" mutations only present in one area.
The lineages of cells in which all these DNA alterations accumulate are difficult to trace, but two recent lines of evidence suggest that normal stem cells may be the cells of origin in cancers. First, there exists a highly positive correlation (Spearman's rho = 0.81; P < 3.5 × 10−8) between the risk of developing cancer in a tissue and the number of normal stem cell divisions taking place in that same tissue. The correlation applied to 31 cancer types and extended across five orders of magnitude. This correlation means that if normal stem cells from a tissue divide once, the cancer risk in that tissue is approximately 1X. If they divide 1,000 times, the cancer risk is 1,000X. And if the normal stem cells from a tissue divide 100,000 times, the cancer risk in that tissue is approximately 100,000X. This strongly suggests that the main factor in cancer initiation is the fact that "normal" stem cells divide, which implies that cancer originates in normal, healthy stem cells.
Second, statistics show that most human cancers are diagnosed in older people. A possible explanation is that cancers occur because cells accumulate damage through time. DNA is the only cellular component that can accumulate damage over the entire course of a life, and stem cells are the only cells that can transmit DNA from the zygote to cells late in life. Other cells, derived from stem cells, do not keep DNA from the beginning of life until a possible cancer occurs. This implies that most cancers arise from normal stem cells.
Contribution of field defects

The term "field cancerization" was first used in 1953 to describe an area or "field" of epithelium that has been preconditioned by (at that time) largely unknown processes so as to predispose it towards development of cancer. Since then, the terms "field cancerization" and "field defect" have been used to describe pre-malignant tissue in which new cancers are likely to arise.
Field defects have been identified in association with cancers and are important in progression to cancer. However, it was pointed out by Rubin that "the vast majority of studies in cancer research has been done on well-defined tumors in vivo, or on discrete neoplastic foci in vitro. Yet there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion…" More than half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of apparently normal cells. It would also be expected that many of the epigenetic alterations present in tumors may have occurred in pre-neoplastic field defects.
In the colon, a field defect probably arises by natural selection of a mutant or epigenetically altered cell among the stem cells at the base of one of the intestinal crypts on the inside surface of the colon. A mutant or epigenetically altered stem cell may replace the other nearby stem cells by natural selection. This may cause a patch of abnormal tissue to arise. The figure in this section includes a photo of a freshly resected and lengthwise-opened segment of the colon showing a colon cancer and four polyps. Below the photo there is a schematic diagram of how a large patch of mutant or epigenetically altered cells may have formed, shown by the large area in yellow in the diagram. Within this first large patch in the diagram (a large clone of cells), a second such mutation or epigenetic alteration may occur, so that a given stem cell acquires an advantage compared to its neighbors, and this altered stem cell may expand clonally, forming a secondary patch, or sub-clone, within the original patch. This is indicated in the diagram by four smaller patches of different colors within the large yellow original area. Within these new patches (sub-clones), the process may be repeated multiple times, indicated by the still smaller patches within the four secondary patches (with still different colors in the diagram) which clonally expand, until stem cells arise that generate either small polyps or else a malignant neoplasm (cancer). In the photo, an apparent field defect in this segment of a colon has generated four polyps (labeled with the size of the polyps, 6mm, 5mm, and two of 3mm, and a cancer about 3 cm across in its longest dimension). These neoplasms are also indicated (in the diagram below the photo) by 4 small tan circles (polyps) and a larger red area (cancer). The cancer in the photo occurred in the cecal area of the colon, where the colon joins the small intestine (labeled) and where the appendix occurs (labeled). The fat in the photo is external to the outer wall of the colon. In the segment of colon shown here, the colon was cut open lengthwise to expose its inner surface and to display the cancer and polyps occurring within the inner epithelial lining of the colon.
If the general process by which sporadic colon cancers arise is the formation of a pre-neoplastic clone that spreads by natural selection, followed by formation of internal sub-clones within the initial clone, and sub-sub-clones inside those, then colon cancers generally should be associated with, and be preceded by, fields of increasing abnormality, reflecting the succession of premalignant events. The most extensive region of abnormality (the outermost yellow irregular area in the diagram) would reflect the earliest event in formation of a malignant neoplasm.
In experimental evaluation of specific DNA repair deficiencies in cancers, many specific DNA repair deficiencies were also shown to occur in the field defects surrounding those cancers. The table below gives examples for which the DNA repair deficiency in a cancer was shown to be caused by an epigenetic alteration, and the somewhat lower frequencies with which the same epigenetically caused DNA repair deficiency was found in the surrounding field defect.
| Cancer | Gene | Frequency in Cancer | Frequency in Field Defect | Reference |
|---|---|---|---|---|
| Colorectal | MGMT | 46% | 34% | |
| Colorectal | MGMT | 47% | 11% | |
| Colorectal | MGMT | 70% | 60% | |
| Colorectal | MSH2 | 13% | 5% | |
| Colorectal | ERCC1 | 100% | 40% | |
| Colorectal | PMS2 | 88% | 50% | |
| Colorectal | XPF | 55% | 40% | |
| Head and Neck | MGMT | 54% | 38% | |
| Head and Neck | MLH1 | 33% | 25% | |
| Head and Neck | MLH1 | 31% | 20% | |
| Stomach | MGMT | 88% | 78% | |
| Stomach | MLH1 | 73% | 20% | |
| Esophagus | MLH1 | 77%–100% | 23%–79% |
Some of the small polyps in the field defect shown in the photo of the opened colon segment may be relatively benign neoplasms. In a 1996 study of polyps less than 10mm in size found during colonoscopy and followed with repeat colonoscopies for 3 years, 25% remained unchanged in size, 35% regressed or shrank in size and 40% grew in size.
Genome instability
Cancers are known to exhibit genome instability or a "mutator phenotype". The protein-coding DNA within the nucleus is about 1.5% of the total genomic DNA. Within this protein-coding DNA (called the exome), an average cancer of the breast or colon can have about 60 to 70 protein altering mutations, of which about 3 or 4 may be "driver" mutations, and the remaining ones may be "passenger" mutations. However, the average number of DNA sequence mutations in the entire genome (including non-protein-coding regions) within a breast cancer tissue sample is about 20,000. In an average melanoma tissue sample (melanomas have a higher exome mutation frequency),) the total number of DNA sequence mutations is about 80,000. These high frequencies of mutations in the total nucleotide sequences within cancers suggest that often an early alteration in the field defect giving rise to a cancer (e.g. yellow area in the diagram in the preceding section) is a deficiency in DNA repair. Large field defects surrounding colon cancers (extending to about 10 cm on each side of a cancer) are found to frequently have epigenetic defects in two or three DNA repair proteins (ERCC1, ERCC4 (XPF) and/or PMS2) in the entire area of the field defect. When expression of DNA repair genes is reduced, DNA damage accumulates in cells at a higher than normal rate, and this excess damage causes an increased frequency of mutation and/or epimutation. Mutation rates strongly increase in cells defective in DNA mismatch repair or in homologous recombinational repair (HRR). A deficiency in DNA repair, itself, can allow DNA damage to accumulate, and error-prone translesion synthesis of some of the damaged areas may give rise to mutations. In addition, faulty repair of this accumulated DNA damage may give rise to epimutations. These new mutations and/or epimutations may provide a proliferative advantage, generating a field defect. Although the mutations/epimutations in DNA repair genes do not, themselves, confer a selective advantage, they may be carried along as passengers in cells when the cell acquires an additional mutation/epimutation that does provide a proliferative advantage.
Non-mainstream theories
There are a number of theories of carcinogenesis and cancer treatment that fall outside the mainstream of scientific opinion, due to lack of scientific rationale, logic, or evidence base. These theories may be used to justify various alternative cancer treatments. They should be distinguished from those theories of carcinogenesis that have a logical basis within mainstream cancer biology, and from which conventionally testable hypotheses can be made.
Several alternative theories of carcinogenesis, however, are based on scientific evidence and are increasingly being acknowledged. Some researchers believe that cancer may be caused by aneuploidy (numerical and structural abnormalities in chromosomes) rather than by mutations or epimutations. Cancer has also been considered as a metabolic disease, in which the cellular metabolism of oxygen is diverted from the pathway that generates energy (oxidative phosphorylation) to the pathway that generates reactive oxygen species. This causes an energy switch from oxidative phosphorylation to aerobic glycolysis (Warburg's hypothesis), and the accumulation of reactive oxygen species leading to oxidative stress ("oxidative stress theory of cancer"). Another concept of cancer development is based on exposure to weak magnetic and electromagnetic fields and their effects on oxidative stress, known as magnetocarcinogenesis.
A number of authors have questioned the assumption that cancers result from sequential random mutations as oversimplistic, suggesting instead that cancer results from a failure of the body to inhibit an innate, programmed proliferative tendency. A related theory suggests that cancer is an atavism, an evolutionary throwback to an earlier form of multicellular life. The genes responsible for uncontrolled cell growth and cooperation between cancer cells are very similar to those that enabled the first multicellular life forms to group together and flourish. These genes still exist within the genomes of more complex metazoans, such as humans, although more recently evolved genes keep them in check. When the newer controlling genes fail for whatever reason, the cell can revert to its more primitive programming and reproduce out of control. The theory is an alternative to the notion that cancers begin with rogue cells that undergo evolution within the body. Instead, they possess a fixed number of primitive genes that are progressively activated, giving them finite variability. Another evolutionary theory puts the roots of cancer back to the origin of the eukaryote (nucleated) cell by massive horizontal gene transfer, when the genomes of infecting viruses were cleaved (and thereby attenuated) by the host, but their fragments integrated into the host genome as immune protection. Cancer thus originates when a rare somatic mutation recombines such fragments into a functional driver of cell proliferation.
Cancer cell biology
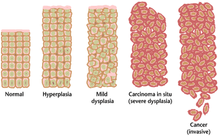
Often, the multiple genetic changes that result in cancer may take many years to accumulate. During this time, the biological behavior of the pre-malignant cells slowly changes from the properties of normal cells to cancer-like properties. Pre-malignant tissue can have a distinctive appearance under the microscope. Among the distinguishing traits of a pre-malignant lesion are an increased number of dividing cells, variation in nuclear size and shape, variation in cell size and shape, loss of specialized cell features, and loss of normal tissue organization. Dysplasia is an abnormal type of excessive cell proliferation characterized by loss of normal tissue arrangement and cell structure in pre-malignant cells. These early neoplastic changes must be distinguished from hyperplasia, a reversible increase in cell division caused by an external stimulus, such as a hormonal imbalance or chronic irritation.
The most severe cases of dysplasia are referred to as carcinoma in situ. In Latin, the term in situ means "in place"; carcinoma in situ refers to an uncontrolled growth of dysplastic cells that remains in its original location and has not shown invasion into other tissues. Carcinoma in situ may develop into an invasive malignancy and is usually removed surgically when detected.
Clonal evolution
Just as a population of animals undergoes evolution, an unchecked population of cells also can undergo "evolution". This undesirable process is called somatic evolution, and is how cancer arises and becomes more malignant over time.
Most changes in cellular metabolism that allow cells to grow in a disorderly fashion lead to cell death. However, once cancer begins, cancer cells undergo a process of natural selection: the few cells with new genetic changes that enhance their survival or reproduction multiply faster, and soon come to dominate the growing tumor as cells with less favorable genetic change are out-competed. This is the same mechanism by which pathogenic species such as MRSA can become antibiotic-resistant and by which HIV can become drug-resistant), and by which plant diseases and insects can become pesticide-resistant. This evolution explains why a cancer relapse often involves cells that have acquired cancer-drug resistance or resistance to radiotherapy).
Biological properties of cancer cells
In a 2000 article by Hanahan and Weinberg, the biological properties of malignant tumor cells were summarized as follows:
- Acquisition of self-sufficiency in growth signals, leading to unchecked growth.
- Loss of sensitivity to anti-growth signals, also leading to unchecked growth.
- Loss of capacity for apoptosis, allowing growth despite genetic errors and external anti-growth signals.
- Loss of capacity for senescence, leading to limitless replicative potential (immortality)
- Acquisition of sustained angiogenesis, allowing the tumor to grow beyond the limitations of passive nutrient diffusion.
- Acquisition of ability to invade neighbouring tissues, the defining property of invasive carcinoma.
- Acquisition of ability to seed metastases at distant sites, a late-appearing property of some malignant tumors (carcinomas or others).
The completion of these multiple steps would be a very rare event without:
- Loss of capacity to repair genetic errors, leading to an increased mutation rate (genomic instability), thus accelerating all the other changes.
These biological changes are classical in carcinomas; other malignant tumors may not need to achieve them all. For example, given that tissue invasion and displacement to distant sites are normal properties of leukocytes, these steps are not needed in the development of leukemia. Nor do the different steps necessarily represent individual mutations. For example, inactivation of a single gene, coding for the p53 protein, will cause genomic instability, evasion of apoptosis and increased angiogenesis. Further, not all the cancer cells are dividing. Rather, a subset of the cells in a tumor, called cancer stem cells, replicate themselves as they generate differentiated cells.
Cancer as a defect in cell interactions
Normally, once a tissue is injured or infected, damaged cells elicit inflammation by stimulating specific patterns of enzyme activity and cytokine gene expression in surrounding cells. Discrete clusters ("cytokine clusters") of molecules are secreted, which act as mediators, inducing the activity of subsequent cascades of biochemical changes. Each cytokine binds to specific receptors on various cell types, and each cell type responds in turn by altering the activity of intracellular signal transduction pathways, depending on the receptors that the cell expresses and the signaling molecules present inside the cell. Collectively, this reprogramming process induces a stepwise change in cell phenotypes, which will ultimately lead to restoration of tissue function and toward regaining essential structural integrity. A tissue can thereby heal, depending on the productive communication between the cells present at the site of damage and the immune system. One key factor in healing is the regulation of cytokine gene expression, which enables complementary groups of cells to respond to inflammatory mediators in a manner that gradually produces essential changes in tissue physiology. Cancer cells have either permanent (genetic) or reversible (epigenetic) changes to their genome, which partly inhibit their communication with surrounding cells and with the immune system. Cancer cells do not communicate with their tissue microenvironment in a manner that protects tissue integrity; instead, the movement and the survival of cancer cells become possible in locations where they can impair tissue function. Cancer cells survive by "rewiring" signal pathways that normally protect the tissue from the immune system. This alteration of the immune response is evident in early stages of malignancy too.
One example of tissue function rewiring in cancer is the activity of transcription factor NF-κB. NF-κB activates the expression of numerous genes involved in the transition between inflammation and regeneration, which encode cytokines, adhesion factors, and other molecules that can change cell fate. This reprogramming of cellular phenotypes normally allows the development of a fully functional intact tissue. NF-κB activity is tightly controlled by multiple proteins, which collectively ensure that only discrete clusters of genes are induced by NF-κB in a given cell and at a given time. This tight regulation of signal exchange between cells protects the tissue from excessive inflammation, and ensures that different cell types gradually acquire complementary functions and specific positions. Failure of this mutual regulation between genetic reprogramming and cell interactions allows cancer cells to give rise to metastasis. Cancer cells respond aberrantly to cytokines, and activate signal cascades that can protect them from the immune system.
In fish
The role of iodine in marine fish (rich in iodine) and freshwater fish (iodine-deficient) is not completely understood, but it has been reported that freshwater fish are more susceptible to infectious and, in particular, neoplastic and atherosclerotic diseases, than marine fish. Marine elasmobranch fishes such as sharks, stingrays etc. are much less affected by cancer than freshwater fishes, and therefore have stimulated medical research to better understand carcinogenesis.
Mechanisms
In order for cells to start dividing uncontrollably, genes that regulate cell growth must be dysregulated.Proto-oncogenes are genes that promote cell growth and mitosis, whereas tumor suppressor genes discourage cell growth, or temporarily halt cell division to carry out DNA repair. Typically, a series of several mutations to these genes is required before a normal cell transforms into a cancer cell. This concept is sometimes termed "oncoevolution." Mutations to these genes provide the signals for tumor cells to start dividing uncontrollably. But the uncontrolled cell division that characterizes cancer also requires that the dividing cell duplicates all its cellular components to create two daughter cells. The activation of anaerobic glycolysis (the Warburg effect), which is not necessarily induced by mutations in proto-oncogenes and tumor suppressor genes, provides most of the building blocks required to duplicate the cellular components of a dividing cell and, therefore, is also essential for carcinogenesis.
Oncogenes
Oncogenes promote cell growth through a variety of ways. Many can produce hormones, a "chemical messenger" between cells that encourage mitosis, the effect of which depends on the signal transduction of the receiving tissue or cells. In other words, when a hormone receptor on a recipient cell is stimulated, the signal is conducted from the surface of the cell to the cell nucleus to affect some change in gene transcription regulation at the nuclear level. Some oncogenes are part of the signal transduction system itself, or the signal receptors in cells and tissues themselves, thus controlling the sensitivity to such hormones. Oncogenes often produce mitogens, or are involved in transcription of DNA in protein synthesis, which creates the proteins and enzymes responsible for producing the products and biochemicals cells use and interact with.
Mutations in proto-oncogenes, which are the normally quiescent counterparts of oncogenes, can modify their expression and function, increasing the amount or activity of the product protein. When this happens, the proto-oncogenes become oncogenes, and this transition upsets the normal balance of cell cycle regulation in the cell, making uncontrolled growth possible. The chance of cancer cannot be reduced by removing proto-oncogenes from the genome, even if this were possible, as they are critical for growth, repair and homeostasis of the organism. It is only when they become mutated that the signals for growth become excessive.
One of the first oncogenes to be defined in cancer research is the ras oncogene. Mutations in the Ras family of proto-oncogenes (comprising H-Ras, N-Ras and K-Ras) are very common, being found in 20% to 30% of all human tumours. Ras was originally identified in the Harvey sarcoma virus genome, and researchers were surprised that not only is this gene present in the human genome but also, when ligated to a stimulating control element, it could induce cancers in cell line cultures.
Proto-oncogenes
Proto-oncogenes promote cell growth in a variety of ways. Many can produce hormones, "chemical messengers" between cells that encourage mitosis, the effect of which depends on the signal transduction of the receiving tissue or cells. Some are responsible for the signal transduction system and signal receptors in cells and tissues themselves, thus controlling the sensitivity to such hormones. They often produce mitogens, or are involved in transcription of DNA in protein synthesis, which create the proteins and enzymes responsible for producing the products and biochemicals cells use and interact with.
Mutations in proto-oncogenes can modify their expression and function, increasing the amount or activity of the product protein. When this happens, they become oncogenes, and, thus, cells have a higher chance of dividing excessively and uncontrollably. The chance of cancer cannot be reduced by removing proto-oncogenes from the genome, as they are critical for growth, repair and homeostasis of the body. It is only when they become mutated that the signals for growth become excessive. It is important to note that a gene possessing a growth-promoting role may increase the carcinogenic potential of a cell, under the condition that all necessary cellular mechanisms that permit growth are activated. This condition also includes the inactivation of specific tumor suppressor genes (see below). If the condition is not fulfilled, the cell may cease to grow and can proceed to die. This makes identification of the stage and type of cancer cell that grows under the control of a given oncogene crucial for the development of treatment strategies.
Tumor suppressor genes
Tumor suppressor genes code for anti-proliferation signals and proteins that suppress mitosis and cell growth. Generally, tumor suppressors are transcription factors that are activated by cellular stress or DNA damage. Often DNA damage will cause the presence of free-floating genetic material as well as other signs, and will trigger enzymes and pathways that lead to the activation of tumor suppressor genes. The functions of such genes is to arrest the progression of the cell cycle in order to carry out DNA repair, preventing mutations from being passed on to daughter cells. The p53 protein, one of the most important studied tumor suppressor genes, is a transcription factor activated by many cellular stressors including hypoxia and ultraviolet radiation damage.
Despite nearly half of all cancers possibly involving alterations in p53, its tumor suppressor function is poorly understood. p53 clearly has two functions: one a nuclear role as a transcription factor, and the other a cytoplasmic role in regulating the cell cycle, cell division, and apoptosis.
The Warburg hypothesis is the preferential use of glycolysis for energy to sustain cancer growth. p53 has been shown to regulate the shift from the respiratory to the glycolytic pathway.
However, a mutation can damage the tumor suppressor gene itself, or the signal pathway that activates it, "switching it off". The invariable consequence of this is that DNA repair is hindered or inhibited: DNA damage accumulates without repair, inevitably leading to cancer.
Mutations of tumor suppressor genes that occur in germline cells are passed along to offspring, and increase the likelihood for cancer diagnoses in subsequent generations. Members of these families have increased incidence and decreased latency of multiple tumors. The tumor types are typical for each type of tumor suppressor gene mutation, with some mutations causing particular cancers, and other mutations causing others. The mode of inheritance of mutant tumor suppressors is that an affected member inherits a defective copy from one parent, and a normal copy from the other. For instance, individuals who inherit one mutant p53 allele (and are therefore heterozygous for mutated p53) can develop melanomas and pancreatic cancer, known as Li-Fraumeni syndrome. Other inherited tumor suppressor gene syndromes include Rb mutations, linked to retinoblastoma, and APC gene mutations, linked to adenopolyposis colon cancer. Adenopolyposis colon cancer is associated with thousands of polyps in colon while young, leading to colon cancer at a relatively early age. Finally, inherited mutations in BRCA1 and BRCA2 lead to early onset of breast cancer.
Development of cancer was proposed in 1971 to depend on at least two mutational events. In what became known as the Knudson two-hit hypothesis, an inherited, germ-line mutation in a tumor suppressor gene would cause cancer only if another mutation event occurred later in the organism's life, inactivating the other allele of that tumor suppressor gene.
Usually, oncogenes are dominant, as they contain gain-of-function mutations, while mutated tumor suppressors are recessive, as they contain loss-of-function mutations. Each cell has two copies of the same gene, one from each parent, and under most cases gain of function mutations in just one copy of a particular proto-oncogene is enough to make that gene a true oncogene. On the other hand, loss of function mutations need to happen in both copies of a tumor suppressor gene to render that gene completely non-functional. However, cases exist in which one mutated copy of a tumor suppressor gene can render the other, wild-type copy non-functional. This phenomenon is called the dominant negative effect and is observed in many p53 mutations.
Knudson's two hit model has recently been challenged by several investigators. Inactivation of one allele of some tumor suppressor genes is sufficient to cause tumors. This phenomenon is called haploinsufficiency and has been demonstrated by a number of experimental approaches. Tumors caused by haploinsufficiency usually have a later age of onset when compared with those by a two hit process.
Multiple mutations

In general, mutations in both types of genes are required for cancer to occur. For example, a mutation limited to one oncogene would be suppressed by normal mitosis control and tumor suppressor genes, first hypothesised by the Knudson hypothesis. A mutation to only one tumor suppressor gene would not cause cancer either, due to the presence of many "backup" genes that duplicate its functions. It is only when enough proto-oncogenes have mutated into oncogenes, and enough tumor suppressor genes deactivated or damaged, that the signals for cell growth overwhelm the signals to regulate it, that cell growth quickly spirals out of control. Often, because these genes regulate the processes that prevent most damage to genes themselves, the rate of mutations increases as one gets older, because DNA damage forms a feedback loop.
Mutation of tumor suppressor genes that are passed on to the next generation of not merely cells, but their offspring, can cause increased likelihoods for cancers to be inherited. Members within these families have increased incidence and decreased latency of multiple tumors. The mode of inheritance of mutant tumor suppressors is that affected member inherits a defective copy from one parent, and a normal copy from another. Because mutations in tumor suppressors act in a recessive manner (note, however, there are exceptions), the loss of the normal copy creates the cancer phenotype. For instance, individuals that are heterozygous for p53 mutations are often victims of Li-Fraumeni syndrome, and that are heterozygous for Rb mutations develop retinoblastoma. In similar fashion, mutations in the adenomatous polyposis coli gene are linked to adenopolyposis colon cancer, with thousands of polyps in the colon while young, whereas mutations in BRCA1 and BRCA2 lead to early onset of breast cancer.
A new idea announced in 2011 is an extreme version of multiple mutations, called chromothripsis by its proponents. This idea, affecting only 2–3% of cases of cancer, although up to 25% of bone cancers, involves the catastrophic shattering of a chromosome into tens or hundreds of pieces and then being patched back together incorrectly. This shattering probably takes place when the chromosomes are compacted during normal cell division, but the trigger for the shattering is unknown. Under this model, cancer arises as the result of a single, isolated event, rather than the slow accumulation of multiple mutations.
Non-mutagenic carcinogens
Many mutagens are also carcinogens, but some carcinogens are not mutagens. Examples of carcinogens that are not mutagens include alcohol and estrogen. These are thought to promote cancers through their stimulating effect on the rate of cell mitosis. Faster rates of mitosis increasingly leave fewer opportunities for repair enzymes to repair damaged DNA during DNA replication, increasing the likelihood of a genetic mistake. A mistake made during mitosis can lead to the daughter cells' receiving the wrong number of chromosomes, which leads to aneuploidy and may lead to cancer.
Role of infections
Bacterial
Helicobacter pylori can cause gastric cancer. Although the data varies between different countries, overall about 1% to 3% of people infected with Helicobacter pylori develop gastric cancer in their lifetime compared to 0.13% of individuals who have had no H. pylori infection.H. pylori infection is very prevalent. As evaluated in 2002, it is present in the gastric tissues of 74% of middle-aged adults in developing countries and 58% in developed countries. Since 1% to 3% of infected individuals are likely to develop gastric cancer,H. pylori-induced gastric cancer is the third highest cause of worldwide cancer mortality as of 2018.
Infection by H. pylori causes no symptoms in about 80% of those infected. About 75% of individuals infected with H. pylori develop gastritis. Thus, the usual consequence of H. pylori infection is chronic asymptomatic gastritis. Because of the usual lack of symptoms, when gastric cancer is finally diagnosed it is often fairly advanced. More than half of gastric cancer patients have lymph node metastasis when they are initially diagnosed.
The gastritis caused by H. pylori is accompanied by inflammation, characterized by infiltration of neutrophils and macrophages to the gastric epithelium, which favors the accumulation of pro-inflammatory cytokines and reactive oxygen species/reactive nitrogen species (ROS/RNS). The substantial presence of ROS/RNS causes DNA damage including 8-oxo-2'-deoxyguanosine (8-OHdG). If the infecting H. pylori carry the cytotoxic cagA gene (present in about 60% of Western isolates and a higher percentage of Asian isolates), they can increase the level of 8-OHdG in gastric cells by 8-fold, while if the H. pylori do not carry the cagA gene, the increase in 8-OHdG is about 4-fold. In addition to the oxidative DNA damage 8-OHdG, H. pylori infection causes other characteristic DNA damages including DNA double-strand breaks.
H. pylori also causes many epigenetic alterations linked to cancer development. These epigenetic alterations are due to H. pylori-induced methylation of CpG sites in promoters of genes and H. pylori-induced altered expression of multiple microRNAs.
As reviewed by Santos and RibeiroH. pylori infection is associated with epigenetically reduced efficiency of the DNA repair machinery, which favors the accumulation of mutations and genomic instability as well as gastric carcinogenesis. In particular, Raza et al. showed that expression of two DNA repair proteins, ERCC1 and PMS2, was severely reduced once H. pylori infection had progressed to cause dyspepsia. Dyspepsia occurs in about 20% of infected individuals. In addition, as reviewed by Raza et al., human gastric infection with H. pylori causes epigenetically reduced protein expression of DNA repair proteins MLH1, MGMT and MRE11. Reduced DNA repair in the presence of increased DNA damage increases carcinogenic mutations and is likely a significant cause of H. pylori carcinogenesis.
Viral
Furthermore, many cancers originate from a viral infection; this is especially true in animals such as birds, but less so in humans. 12% of human cancers can be attributed to a viral infection. The mode of virally induced tumors can be divided into two, acutely transforming or slowly transforming. In acutely transforming viruses, the viral particles carry a gene that encodes for an overactive oncogene called viral-oncogene (v-onc), and the infected cell is transformed as soon as v-onc is expressed. In contrast, in slowly transforming viruses, the virus genome is inserted, especially as viral genome insertion is an obligatory part of retroviruses, near a proto-oncogene in the host genome. The viral promoter or other transcription regulation elements, in turn, cause over-expression of that proto-oncogene, which, in turn, induces uncontrolled cellular proliferation. Because viral genome insertion is not specific to proto-oncogenes and the chance of insertion near that proto-oncogene is low, slowly transforming viruses have very long tumor latency compared to acutely transforming virus, which already carries the viral-oncogene.
Viruses that are known to cause cancer such as HPV (cervical cancer), Hepatitis B (liver cancer), and EBV (a type of lymphoma), are all DNA viruses. It is thought that when the virus infects a cell, it inserts a part of its own DNA near the cell growth genes, causing cell division. The group of changed cells that are formed from the first cell dividing all have the same viral DNA near the cell growth genes. The group of changed cells are now special because one of the normal controls on growth has been lost.
Depending on their location, cells can be damaged through radiation, chemicals from cigarette smoke, and inflammation from bacterial infection or other viruses. Each cell has a chance of damage. Cells often die if they are damaged, through failure of a vital process or the immune system, however, sometimes damage will knock out a single cancer gene. In an old person, there are thousands, tens of thousands, or hundreds of thousands of knocked-out cells. The chance that any one would form a cancer is very low.
When the damage occurs in any area of changed cells, something different occurs. Each of the cells has the potential for growth. The changed cells will divide quicker when the area is damaged by physical, chemical, or viral agents. A vicious circle has been set up: Damaging the area will cause the changed cells to divide, causing a greater likelihood that they will experience knock-outs.
This model of carcinogenesis is popular because it explains why cancers grow. It would be expected that cells that are damaged through radiation would die or at least be worse off because they have fewer genes working; viruses increase the number of genes working.
One thought is that we may end up with thousands of vaccines to prevent every virus that can change our cells. Viruses can have different effects on different parts of the body. It may be possible to prevent a number of different cancers by immunizing against one viral agent. It is likely that HPV, for instance, has a role in cancers of the mucous membranes of the mouth.
Helminthiasis
Certain parasitic worms are known to be carcinogenic. These include:
- Clonorchis sinensis (the organism causing Clonorchiasis) and Opisthorchis viverrini (causing Opisthorchiasis) are associated with cholangiocarcinoma.
- Schistosoma species (the organisms causing Schistosomiasis) is associated with bladder cancer.
Epigenetics
Epigenetics is the study of the regulation of gene expression through chemical, non-mutational changes in DNA structure. The theory of epigenetics in cancer pathogenesis is that non-mutational changes to DNA can lead to alterations in gene expression. Normally, oncogenes are silent, for example, because of DNA methylation. Loss of that methylation can induce the aberrant expression of oncogenes, leading to cancer pathogenesis. Known mechanisms of epigenetic change include DNA methylation, and methylation or acetylation of histone proteins bound to chromosomal DNA at specific locations. Classes of medications, known as HDAC inhibitors and DNA methyltransferase inhibitors, can re-regulate the epigenetic signaling in the cancer cell.
Epimutations include methylations or demethylations of the CpG islands of the promoter regions of genes, which result in repression or de-repression, respectively of gene expression. Epimutations can also occur by acetylation, methylation, phosphorylation or other alterations to histones, creating a histone code that represses or activates gene expression, and such histone epimutations can be important epigenetic factors in cancer. In addition, carcinogenic epimutation can occur through alterations of chromosome architecture caused by proteins such as HMGA2. A further source of epimutation is due to increased or decreased expression of microRNAs (miRNAs). For example, extra expression of miR-137 can cause downregulation of expression of 491 genes, and miR-137 is epigenetically silenced in 32% of colorectal cancers>
Cancer stem cells
A new way of looking at carcinogenesis comes from integrating the ideas of developmental biology into oncology. The cancer stem cell hypothesis proposes that the different kinds of cells in a heterogeneous tumor arise from a single cell, termed Cancer Stem Cell. Cancer stem cells may arise from transformation of adult stem cells or differentiated cells within a body. These cells persist as a subcomponent of the tumor and retain key stem cell properties. They give rise to a variety of cells, are capable of self-renewal and homeostatic control. Furthermore, the relapse of cancer and the emergence of metastasis are also attributed to these cells. The cancer stem cell hypothesis does not contradict earlier concepts of carcinogenesis. The cancer stem cell hypothesis has been a proposed mechanism that contributes to tumour heterogeneity.
Clonal evolution
While genetic and epigenetic alterations in tumor suppressor genes and oncogenes change the behavior of cells, those alterations, in the end, result in cancer through their effects on the population of neoplastic cells and their microenvironment. Mutant cells in neoplasms compete for space and resources. Thus, a clone with a mutation in a tumor suppressor gene or oncogene will expand only in a neoplasm if that mutation gives the clone a competitive advantage over the other clones and normal cells in its microenvironment. Thus, the process of carcinogenesis is formally a process of Darwinian evolution, known as somatic or clonal evolution. Furthermore, in light of the Darwinistic mechanisms of carcinogenesis, it has been theorized that the various forms of cancer can be categorized as pubertal and gerontological. Anthropological research is currently being conducted on cancer as a natural evolutionary process through which natural selection destroys environmentally inferior phenotypes while supporting others. According to this theory, cancer comes in two separate types: from birth to the end of puberty (approximately age 20) teleologically inclined toward supportive group dynamics, and from mid-life to death (approximately age 40+) teleologically inclined away from overpopulated group dynamics.
See also
- Cancer cell
- Armitage–Doll multistage model of carcinogenesis
- Linear no-threshold model
- Oncogenic retroviridae protein
Further reading
- Tokar EJ, Benbrahim-Tallaa L, Waalkes MP (2011). "Chepter 14. Metal Ions in Human Cancer Development". In Sigel A, Sigel H, Sigel RK (eds.). Metal ions in toxicology: effects, interactions, interdependencies. Metal Ions in Life Sciences. Vol. 8. RSC Publishing. pp. 375–401. doi:10.1039/9781849732116-00375. ISBN 978-1-84973-091-4.
- Dixon K, Kopras E (December 2004). "Genetic alterations and DNA repair in human carcinogenesis". Seminars in Cancer Biology. 14 (6): 441–8. doi:10.1016/j.semcancer.2004.06.007. PMID 15489137.
- Kleinsmith LJ (2006). Principles of cancer biology. San Francisco: Pearson Benjamin Cummings. ISBN 978-0-8053-4003-7.
- Sarasin A (November 2003). "An overview of the mechanisms of mutagenesis and carcinogenesis". Mutation Research. 544 (2–3): 99–106. doi:10.1016/j.mrrev.2003.06.024. PMID 14644312.
- Schottenfeld D, Beebe-Dimmer JL (2005). "Advances in cancer epidemiology: understanding causal mechanisms and the evidence for implementing interventions". Annual Review of Public Health. 26: 37–60. doi:10.1146/annurev.publhealth.26.021304.144402. PMID 15760280.
- Tannock I, Hill R, Bristow R, Harrington L (2005). The basic science of oncology (4th ed.). New York: McGraw-Hill. ISBN 978-0-07-138774-3.
- Wicha MS, Liu S, Dontu G (February 2006). "Cancer stem cells: an old idea--a paradigm shift". Cancer Research. 66 (4): 1883–90, discussion 1895–6. doi:10.1158/0008-5472.CAN-05-3153. PMID 16488983.