
Cell cycle analysis by DNA content measurement is a method that most frequently employs flow cytometry to distinguish cells in different phases of the cell cycle. Before analysis, the cells are usually permeabilised and treated with a fluorescent dye that stains DNA quantitatively, such as propidium iodide (PI) or 4,6-diamidino-2-phenylindole (DAPI). The fluorescence intensity of the stained cells correlates with the amount of DNA they contain. As the DNA content doubles during the S phase, the DNA content (and thereby intensity of fluorescence) of cells in the G0 phase and G1 phase (before S), in the S phase, and in the G2 phase and M phase (after S) identifies the cell cycle phase position in the major phases (G0/G1 versus S versus G2/M phase) of the cell cycle. The cellular DNA content of individual cells is often plotted as their frequency histogram to provide information about relative frequency (percentage) of cells in the major phases of the cell cycle.
Cell cycle anomalies revealed on the DNA content frequency histogram are often observed after different types of cell damage, for example such DNA damage that interrupts the cell cycle progression at certain checkpoints. Such an arrest of the cell cycle progression can lead either to an effective DNA repair, which may prevent transformation of normal into a cancer cell (carcinogenesis), or to cell death, often by the mode of apoptosis. An arrest of cells in G0 or G1 is often seen as a result of lack of nutrients (growth factors), for example after serum deprivation. Cell cycle analysis was first described in 1969 at Los Alamos Scientific Laboratory by a group from the University of California using the Feulgen staining technique. The first protocol for cell cycle analysis using propidium iodide staining was presented in 1975 by Awtar Krishan from Harvard Medical School and is still widely cited today.
Multiparameter analysis of the cell cycle includes, in addition to measurement of cellular DNA content, other cell cycle related constituents/features. The concurrent measurement of cellular DNA and RNA content, or DNA susceptibility to denaturation at low pH using the metachromatic dye acridine orange, reveals the G1Q, G1A, and G1B cell cycle compartments and also makes it possible to discriminate between S, G2 and mitotic cells. The cells in G1Q are quiescent, temporarily withdrawn from the cell cycle (also identifiable as G0), the G1A are in the growth phase while G1B are the cells just prior entering S, with their growth (RNA and protein content, size) similar to that of the cells initiating DNA replication. Similar cell cycle compartments are also recognized by multiparameter analysis that includes measurement of expression of cyclin D1, cyclin E, cyclin A and cyclin B1, each in relation to DNA content Concurrent measurement of DNA content and of incorporation of DNA precursor 5-bromo-2'-deoxyuridine (BrdU) by flow cytometry is an especially useful assay, that has been widely used in analysis of the cell cycle in vitro and in vivo. However, the incorporation of 5-ethynyl-2'-deoxyuridine (EdU), the precursor whose detection offers certain advantages over BrdU, has now become the preferred methodology do detect DNA replicating (S-phase) cells.
Experimental procedure

Unless staining is performed using Hoechst 33342, the first step in preparing cells for cell cycle analysis is permeabilisation of the cells' plasma membranes. This is usually done by incubating them in a buffer solution containing a mild detergent such as Triton X-100 or NP-40, or by fixating them in ethanol. Most fluorescent DNA dyes (one of exceptions is Hoechst 33342) are not plasma membrane permeant, that is, unable to pass through an intact cell membrane. Permeabilisation is therefore crucial for the success of the next step, the staining of the cells.
Prior to (or during the staining step) the cells are often treated with RNase A to remove RNAs. This is important because certain dyes that stain DNA will also stain RNA, thus creating artefacts that would distort the results. An exception is the metachromatic fluorochrome acridine orange, which under the specific staining protocol can differentially stain both, RNA (generating red luminescence) and DNA (green fluorescence), or in another protocol, after removal of RNA and partial DNA denaturation, to differentially stain double-stranded DNA (green fluorescence) versus single-stranded DNA (red luminescence)[3]. Aside from propidium iodide and acridine orange, quantifiable dyes that are frequently used include (but are not limited to) DRAQ5, 7-Aminoactinomycin D, DAPI and Hoechst 33342.
Doublet discrimination
Since cells and especially fixed cells tend to stick together, cell aggregates have to be excluded from analysis through a process called doublet discrimination. This is important because a doublet of two G0/G1 cells has the same total content of DNA and thus the same fluorescence intensity as a single G2/M cell. Unless recognized as such the G0/G1 doublets would contribute to false positive identification and count of G2/M cells.
Related methods
Nicoletti assay
The Nicoletti assay, named after its inventor, the Italian physician Ildo Nicoletti, is a modified form of cell cycle analysis. It is used to detect and quantify apoptosis, a form of programmed cell death, by analysing cells with a DNA content less than 2n ("sub-G0/G1 cells"). Such cells are usually the result of apoptotic DNA fragmentation: during apoptosis, the DNA is degraded by cellular endonucleases. Therefore, nuclei of apoptotic cells contain less DNA than nuclei of healthy G0/G1 cells, resulting in a sub-G0/G1 peak in the fluorescence histogram that can be used to determine the relative amount of apoptotic cells in a sample. This method was developed and first described in 1991 by Nicoletti and co-workers at Perugia University School of Medicine. An optimised protocol developed by two of the authors of the original publication was published in 2006. The objects measured within the sub-G0/G1 peak, with DNA content lesser than 5% of that of the G0G1 peak, in all probability are apoptotic bodies and thus do not represent individual apoptotic cells
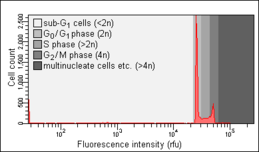
Healthy cells. Note the absence of a sub-G0/G1 peak.

Apoptotic cells one day after apoptosis induction. Note the presence of a sub-G0/G1 peak.

Apoptotic cells several days after apoptosis induction. Note the relative increase of the sub-G0/G1 peak.



Further reading
- "Cell Cycle Basics" (PDF). University College London. Archived from the original (PDF, 0.1 MB) on 2011-06-06. Retrieved 2010-05-20.
- Rabinovitch, Peter. "Introduction to Cell Cycle Analysis" (PDF, 0.5 MB). Phoenix Flow Systems, Inc. Retrieved 2010-05-20.