
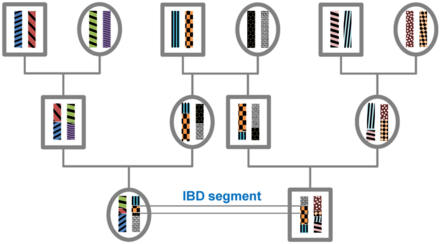
A DNA segment is identical by state (IBS) in two or more individuals if they have identical nucleotide sequences in this segment. An IBS segment is identical by descent (IBD) in two or more individuals if they have inherited it from a common ancestor without recombination, that is, the segment has the same ancestral origin in these individuals. DNA segments that are IBD are IBS per definition, but segments that are not IBD can still be IBS due to the same mutations in different individuals or recombinations that do not alter the segment.

Theory
All individuals in a finite population are related if traced back long enough and will, therefore, share segments of their genomes IBD. During meiosis segments of IBD are broken up by recombination. Therefore, the expected length of an IBD segment depends on the number of generations since the most recent common ancestor at the locus of the segment. The length of IBD segments that result from a common ancestor n generations in the past (therefore involving 2n meiosis) is exponentially distributed with mean 1/(2n) Morgans (M). The expected number of IBD segments decreases with the number of generations since the common ancestor at this locus. For a specific DNA segment, the probability of being IBD decreases as 2−2n since in each meiosis the probability of transmitting this segment is 1/2.
Applications
Identified IBD segments can be used for a wide range of purposes. As noted above the amount (length and number) of IBD sharing depends on the familial relationships between the tested individuals. Therefore, one application of IBD segment detection is to quantify relatedness. Measurement of relatedness can be used in forensic genetics, but can also increase information in genetic linkage mapping and help to decrease bias by undocumented relationships in standard association studies. Another application of IBD is genotype imputation and haplotype phase inference. Long shared segments of IBD, which are broken up by short regions may be indicative for phasing errors.
IBD mapping
IBD mapping is similar to linkage analysis, but can be performed without a known pedigree on a cohort of unrelated individuals. IBD mapping can be seen as a new form of association analysis that increases the power to map genes or genomic regions containing multiple rare disease susceptibility variants.
Using simulated data, Browning and Thompson showed that IBD mapping has higher power than association testing when multiple rare variants within a gene contribute to disease susceptibility. Via IBD mapping, genome-wide significant regions in isolated populations as well as outbred populations were found while standard association tests failed. Houwen et al. used IBD sharing to identify the chromosomal location of a gene responsible for benign recurrent intrahepatic cholestasis in an isolated fishing population. Kenny et al. also used an isolated population to fine-map a signal found by a genome-wide association study (GWAS) of plasma plant sterol (PPS) levels, a surrogate measure of cholesterol absorption from the intestine. Francks et al. was able to identify a potential susceptibility locus for schizophrenia and bipolar disorder with genotype data of case-control samples. Lin et al. found a genome-wide significant linkage signal in a dataset of multiple sclerosis patients. Letouzé et al. used IBD mapping to look for founder mutations in cancer samples.
IBD in population genetics
Detection of natural selection in the human genome is also possible via detected IBD segments. Selection will usually tend to increase the number of IBD segments among individuals in a population. By scanning for regions with excess IBD sharing, regions in the human genome that have been under strong, very recent selection can be identified.
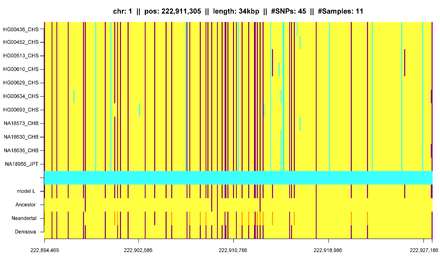
In addition to that, IBD segments can be useful for measuring and identifying other influences on population structure. Gusev et al. showed that IBD segments can be used with additional modeling to estimate demographic history including bottlenecks and admixture. Using similar models Palamara et al. and Carmi et al. reconstructed the demographic history of Ashkenazi Jewish and Kenyan Maasai individuals. Botigué et al. investigated differences in African ancestry among European populations. Ralph and Coop used IBD detection to quantify the common ancestry of different European populations and Gravel et al. similarly tried to draw conclusions of the genetic history of populations in the Americas. Ringbauer et al. utilized geographic structure of IBD segments to estimate dispersal within Eastern Europe during the last centuries. Using the 1000 Genomes data Hochreiter found differences in IBD sharing between African, Asian and European populations as well as IBD segments that are shared with ancient genomes like the Neanderthal or Denisova.
Methods and software
Programs for the detection of IBD segments in unrelated individuals:
- RAPID: Ultra-fast Identity by Descent Detection in Biobank-Scale Cohorts using Positional Burrows–Wheeler Transform
- Parente: identifies IBD segments between pairs of individuals in unphased genotype data
- BEAGLE/fastIBD: finds segments of IBD between pairs of individuals in genome-wide SNP data
- BEAGLE/RefinedIBD: finds IBD segments in pairs of individuals using a hashing method and evaluates their significance via a likelihood ratio
- IBDseq: detects pairwise IBD segments in sequencing data
- GERMLINE: discovers in linear-time IBD segments in pairs of individuals
- DASH: builds upon pairwise IBD segments to infer clusters of individuals likely to be sharing a single haplotype
- PLINK: is a tool set for whole genome association and population-based linkage analyses including a method for pairwise IBD segment detection
- Relate: estimates the probability of IBD between pairs of individuals at a specific locus using SNPs
- MCMC_IBDfinder: is based on Markov chain Monte Carlo (MCMC) for finding IBD segments in multiple individuals
- IBD-Groupon: detects group-wise IBD segments based on pairwise IBD relationships
- HapFABIA: identifies very short IBD segments characterized by rare variants in large sequencing data simultaneously in multiple individuals