
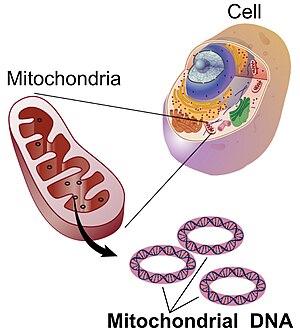
Mitochondrial DNA (mtDNA or mDNA) is the DNA located in mitochondria, cellular organelles within eukaryotic cells that convert chemical energy from food into a form that cells can use, such as adenosine triphosphate (ATP). Mitochondrial DNA is only a small portion of the DNA in a eukaryotic cell; most of the DNA can be found in the cell nucleus and, in plants and algae, also in plastids such as chloroplasts.
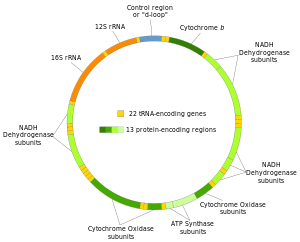
Human mitochondrial DNA was the first significant part of the human genome to be sequenced. This sequencing revealed that the human mtDNA includes 16,569 base pairs and encodes 13 proteins.
Since animal mtDNA evolves faster than nuclear genetic markers, it represents a mainstay of phylogenetics and evolutionary biology. It also permits an examination of the relatedness of populations, and so has become important in anthropology and biogeography.
Origin
Nuclear and mitochondrial DNA are thought to be of separate evolutionary origin, with the mtDNA being derived from the circular genomes of bacteria engulfed by the early ancestors of today's eukaryotic cells. This theory is called the endosymbiotic theory. In the cells of extant organisms, the vast majority of the proteins present in the mitochondria (numbering approximately 1500 different types in mammals) are coded for by nuclear DNA, but the genes for some, if not most, of them are thought to have originally been of bacterial origin, having since been transferred to the eukaryotic nucleus during evolution.
The reasons mitochondria have retained some genes are debated. The existence in some species of mitochondrion-derived organelles lacking a genome suggests that complete gene loss is possible, and transferring mitochondrial genes to the nucleus has several advantages. The difficulty of targeting remotely-produced hydrophobic protein products to the mitochondrion is one hypothesis for why some genes are retained in mtDNA;colocalisation for redox regulation is another, citing the desirability of localised control over mitochondrial machinery. Recent analysis of a wide range of mtDNA genomes suggests that both these features may dictate mitochondrial gene retention.
Genome structure and diversity
Across all organisms, there are six main genome types found in mitochondrial genomes, classified by their structure (i.e. circular versus linear), size, presence of introns or plasmid like structures, and whether the genetic material is a singular molecule or collection of homogeneous or heterogeneous molecules.
In many unicellular organisms (e.g., the ciliate Tetrahymena and the green alga Chlamydomonas reinhardtii), and in rare cases also in multicellular organisms (e.g. in some species of Cnidaria), the mtDNA is found as linearly organized DNA. Most of these linear mtDNAs possess telomerase-independent telomeres (i.e., the ends of the linear DNA) with different modes of replication, which have made them interesting objects of research because many of these unicellular organisms with linear mtDNA are known pathogens.
Animals
Most animals, specifically bilaterian animals, have a circular mitochondrial genome. Medusozoa and calcarea clades however have species with linear mitochondrial chromosomes.
In terms of base pairs, the anemone Isarachnanthus nocturnus has the largest mitochondrial genome of any animal at 80,923 bp. The smallest known mitochondrial genome in animals belongs to the comb jelly Vallicula multiformis, which consist of 9,961 bp.
In February 2020, a jellyfish-related parasite – Henneguya salminicola – was discovered that lacks mitochondrial genome but retains structures deemed mitochondrion-related organelles. Moreover, nuclear DNA genes involved in aerobic respiration and in mitochondrial DNA replication and transcription were either absent or present only as pseudogenes. This is the first multicellular organism known to have this absence of aerobic respiration and lives completely free of oxygen dependency.
Plants and fungi
There are three different mitochondrial genome types found in plants and fungi. The first type is a circular genome that has introns (type 2) and may range from 19 to 1000 kbp in length. The second genome type is a circular genome (about 20–1000 kbp) that also has a plasmid-like structure (1 kb) (type 3). The final genome type that can be found in plants and fungi is a linear genome made up of homogeneous DNA molecules (type 5).
Great variation in mtDNA gene content and size exists among fungi and plants, although there appears to be a core subset of genes that are present in all eukaryotes (except for the few that have no mitochondria at all). In Fungi, however, there is no single gene shared among all mitogenomes. Some plant species have enormous mitochondrial genomes, with Silene conica mtDNA containing as many as 11,300,000 base pairs. Surprisingly, even those huge mtDNAs contain the same number and kinds of genes as related plants with much smaller mtDNAs. The genome of the mitochondrion of the cucumber (Cucumis sativus) consists of three circular chromosomes (lengths 1556, 84 and 45 kilobases), which are entirely or largely autonomous with regard to their replication.
Protists
Protists contain the most diverse mitochondrial genomes, with five different types found in this kingdom. Type 2, type 3 and type 5 mentioned in the plant and fungal genomes also exist in some protists, as do two unique genome types. One of these unique types is a heterogeneous collection of circular DNA molecules (type 4) while the other is a heterogeneous collection of linear molecules (type 6). Genome types 4 and 6 each range from 1–200 kbp in size.
The smallest mitochondrial genome sequenced to date is the 5,967 bp mtDNA of the parasite Plasmodium falciparum.
Endosymbiotic gene transfer, the process by which genes that were coded in the mitochondrial genome are transferred to the cell's main genome, likely explains why more complex organisms such as humans have smaller mitochondrial genomes than simpler organisms such as protists.
| Genome Type | Kingdom | Introns | Size | Shape | Description |
|---|---|---|---|---|---|
| 1 | Animal | No | 11–28 kbp | Circular | Single molecule |
| 2 | Fungi, Plant, Protista | Yes | 19–1000 kbp | Circular | Single molecule |
| 3 | Fungi, Plant, Protista | No | 20–1000 kbp | Circular | Large molecule and small plasmid like structures |
| 4 | Protista | No | 1–200 kbp | Circular | Heterogeneous group of molecules |
| 5 | Fungi, Plant, Protista | No | 1–200 kbp | Linear | Homogeneous group of molecules |
| 6 | Protista | No | 1–200 kbp | Linear | Heterogeneous group of molecules |
Replication
Mitochondrial DNA is replicated by the DNA polymerase gamma complex which is composed of a 140 kDa catalytic DNA polymerase encoded by the POLG gene and two 55 kDa accessory subunits encoded by the POLG2 gene. The replisome machinery is formed by DNA polymerase, TWINKLE and mitochondrial SSB proteins. TWINKLE is a helicase, which unwinds short stretches of dsDNA in the 5' to 3' direction. All these polypeptides are encoded in the nuclear genome.
During embryogenesis, replication of mtDNA is strictly down-regulated from the fertilized oocyte through the preimplantation embryo. The resulting reduction in per-cell copy number of mtDNA plays a role in the mitochondrial bottleneck, exploiting cell-to-cell variability to ameliorate the inheritance of damaging mutations. According to Justin St. John and colleagues, "At the blastocyst stage, the onset of mtDNA replication is specific to the cells of the trophectoderm. In contrast, the cells of the inner cell mass restrict mtDNA replication until they receive the signals to differentiate to specific cell types."

Genes on the human mtDNA and their transcription

The two strands of the human mitochondrial DNA are distinguished as the heavy strand and the light strand. The heavy strand is rich in guanine and encodes 12 subunits of the oxidative phosphorylation system, two ribosomal RNAs (12S and 16S), and 14 transfer RNAs (tRNAs). The light strand encodes one subunit, and 8 tRNAs. So, altogether mtDNA encodes for two rRNAs, 22 tRNAs, and 13 protein subunits, all of which are involved in the oxidative phosphorylation process.
| Gene | Type | Product | Positions in the mitogenome |
Strand |
|---|---|---|---|---|
| MT-ATP8 | protein coding | ATP synthase, Fo subunit 8 (complex V) | 08,366–08,572 (overlap with MT-ATP6) | H |
| MT-ATP6 | protein coding | ATP synthase, Fo subunit 6 (complex V) | 08,527–09,207 (overlap with MT-ATP8) | H |
| MT-CO1 | protein coding | Cytochrome c oxidase, subunit 1 (complex IV) | 05,904–07,445 | H |
| MT-CO2 | protein coding | Cytochrome c oxidase, subunit 2 (complex IV) | 07,586–08,269 | H |
| MT-CO3 | protein coding | Cytochrome c oxidase, subunit 3 (complex IV) | 09,207–09,990 | H |
| MT-CYB | protein coding | Cytochrome b (complex III) | 14,747–15,887 | H |
| MT-ND1 | protein coding | NADH dehydrogenase, subunit 1 (complex I) | 03,307–04,262 | H |
| MT-ND2 | protein coding | NADH dehydrogenase, subunit 2 (complex I) | 04,470–05,511 | H |
| MT-ND3 | protein coding | NADH dehydrogenase, subunit 3 (complex I) | 10,059–10,404 | H |
| MT-ND4L | protein coding | NADH dehydrogenase, subunit 4L (complex I) | 10,470–10,766 (overlap with MT-ND4) | H |
| MT-ND4 | protein coding | NADH dehydrogenase, subunit 4 (complex I) | 10,760–12,137 (overlap with MT-ND4L) | H |
| MT-ND5 | protein coding | NADH dehydrogenase, subunit 5 (complex I) | 12,337–14,148 | H |
| MT-ND6 | protein coding | NADH dehydrogenase, subunit 6 (complex I) | 14,149–14,673 | L |
| MT-RNR2 | protein coding | Humanin | — | — |
| MT-TA | transfer RNA | tRNA-Alanine (Ala or A) | 05,587–05,655 | L |
| MT-TR | transfer RNA | tRNA-Arginine (Arg or R) | 10,405–10,469 | H |
| MT-TN | transfer RNA | tRNA-Asparagine (Asn or N) | 05,657–05,729 | L |
| MT-TD | transfer RNA | tRNA-Aspartic acid (Asp or D) | 07,518–07,585 | H |
| MT-TC | transfer RNA | tRNA-Cysteine (Cys or C) | 05,761–05,826 | L |
| MT-TE | transfer RNA | tRNA-Glutamic acid (Glu or E) | 14,674–14,742 | L |
| MT-TQ | transfer RNA | tRNA-Glutamine (Gln or Q) | 04,329–04,400 | L |
| MT-TG | transfer RNA | tRNA-Glycine (Gly or G) | 09,991–10,058 | H |
| MT-TH | transfer RNA | tRNA-Histidine (His or H) | 12,138–12,206 | H |
| MT-TI | transfer RNA | tRNA-Isoleucine (Ile or I) | 04,263–04,331 | H |
| MT-TL1 | transfer RNA | tRNA-Leucine (Leu-UUR or L) | 03,230–03,304 | H |
| MT-TL2 | transfer RNA | tRNA-Leucine (Leu-CUN or L) | 12,266–12,336 | H |
| MT-TK | transfer RNA | tRNA-Lysine (Lys or K) | 08,295–08,364 | H |
| MT-TM | transfer RNA | tRNA-Methionine (Met or M) | 04,402–04,469 | H |
| MT-TF | transfer RNA | tRNA-Phenylalanine (Phe or F) | 00,577–00,647 | H |
| MT-TP | transfer RNA | tRNA-Proline (Pro or P) | 15,956–16,023 | L |
| MT-TS1 | transfer RNA | tRNA-Serine (Ser-UCN or S) | 07,446–07,514 | L |
| MT-TS2 | transfer RNA | tRNA-Serine (Ser-AGY or S) | 12,207–12,265 | H |
| MT-TT | transfer RNA | tRNA-Threonine (Thr or T) | 15,888–15,953 | H |
| MT-TW | transfer RNA | tRNA-Tryptophan (Trp or W) | 05,512–05,579 | H |
| MT-TY | transfer RNA | tRNA-Tyrosine (Tyr or Y) | 05,826–05,891 | L |
| MT-TV | transfer RNA | tRNA-Valine (Val or V) | 01,602–01,670 | H |
| MT-RNR1 | ribosomal RNA | Small subunit : SSU (12S) | 00,648–01,601 | H |
| MT-RNR2 | ribosomal RNA | Large subunit : LSU (16S) | 01,671–03,229 | H |
Between most (but not all) protein-coding regions, tRNAs are present (see the human mitochondrial genome map). During transcription, the tRNAs acquire their characteristic L-shape that gets recognized and cleaved by specific enzymes. With the mitochondrial RNA processing, individual mRNA, rRNA, and tRNA sequences are released from the primary transcript. Folded tRNAs therefore act as secondary structure punctuations.
Regulation of transcription
The promoters for the initiation of the transcription of the heavy and light strands are located in the main non-coding region of the mtDNA called the displacement loop, the D-loop. There is evidence that the transcription of the mitochondrial rRNAs is regulated by the heavy-strand promoter 1 (HSP1), and the transcription of the polycistronic transcripts coding for the protein subunits are regulated by HSP2.
Measurement of the levels of the mtDNA-encoded RNAs in bovine tissues has shown that there are major differences in the expression of the mitochondrial RNAs relative to total tissue RNA. Among the 12 tissues examined the highest level of expression was observed in heart, followed by brain and steroidogenic tissue samples.
As demonstrated by the effect of the trophic hormone ACTH on adrenal cortex cells, the expression of the mitochondrial genes may be strongly regulated by external factors, apparently to enhance the synthesis of mitochondrial proteins necessary for energy production. Interestingly, while the expression of protein-encoding genes was stimulated by ACTH, the levels of the mitochondrial 16S rRNA showed no significant change.
Mitochondrial inheritance
In most multicellular organisms, mtDNA is inherited from the mother (maternally inherited). Mechanisms for this include simple dilution (an egg contains on average 200,000 mtDNA molecules, whereas a healthy human sperm has been reported to contain on average 5 molecules), degradation of sperm mtDNA in the male genital tract and in the fertilized egg; and, at least in a few organisms, failure of sperm mtDNA to enter the egg. Whatever the mechanism, this single parent (uniparental inheritance) pattern of mtDNA inheritance is found in most animals, most plants and also in fungi.
In a study published in 2018, human babies were reported to inherit mtDNA from both their fathers and their mothers resulting in mtDNA heteroplasmy.
Female inheritance
In sexual reproduction, mitochondria are normally inherited exclusively from the mother; the mitochondria in mammalian sperm are usually destroyed by the egg cell after fertilization. Also, mitochondria are only in the sperm tail, which is used for propelling the sperm cells and sometimes the tail is lost during fertilization. In 1999 it was reported that paternal sperm mitochondria (containing mtDNA) are marked with ubiquitin to select them for later destruction inside the embryo. Some in vitro fertilization techniques, particularly injecting a sperm into an oocyte, may interfere with this.
The fact that mitochondrial DNA is mostly maternally inherited enables genealogical researchers to trace maternal lineage far back in time. (Y-chromosomal DNA, paternally inherited, is used in an analogous way to determine the patrilineal history.) This is usually accomplished on human mitochondrial DNA by sequencing the hypervariable control regions (HVR1 or HVR2), and sometimes the complete molecule of the mitochondrial DNA, as a genealogical DNA test. HVR1, for example, consists of about 440 base pairs. These 440 base pairs are compared to the same regions of other individuals (either specific people or subjects in a database) to determine maternal lineage. Most often, the comparison is made with the revised Cambridge Reference Sequence. Vilà et al. have published studies tracing the matrilineal descent of domestic dogs from wolves. The concept of the Mitochondrial Eve is based on the same type of analysis, attempting to discover the origin of humanity by tracking the lineage back in time.
The mitochondrial bottleneck
Entities subject to uniparental inheritance and with little to no recombination may be expected to be subject to Muller's ratchet, the accumulation of deleterious mutations until functionality is lost. Animal populations of mitochondria avoid this through a developmental process known as the mtDNA bottleneck. The bottleneck exploits random processes in the cell to increase the cell-to-cell variability in mutant load as an organism develops: a single egg cell with some proportion of mutant mtDNA thus produces an embryo in which different cells have different mutant loads. Cell-level selection may then act to remove those cells with more mutant mtDNA, leading to a stabilisation or reduction in mutant load between generations. The mechanism underlying the bottleneck is debated, with a recent mathematical and experimental metastudy providing evidence for a combination of the random partitioning of mtDNAs at cell divisions and the random turnover of mtDNA molecules within the cell.
Male inheritance
Male mitochondrial DNA inheritance has been discovered in Plymouth Rock chickens. Evidence supports rare instances of male mitochondrial inheritance in some mammals as well. Specifically, documented occurrences exist for mice, where the male-inherited mitochondria were subsequently rejected. It has also been found in sheep, and in cloned cattle. Rare cases of male mitochondrial inheritance have been documented in humans. Although many of these cases involve cloned embryos or subsequent rejection of the paternal mitochondria, others document in vivo inheritance and persistence under lab conditions.
Doubly uniparental inheritance of mtDNA is observed in bivalve mollusks. In those species, females have only one type of mtDNA (F), whereas males have F type mtDNA in their somatic cells, but M type of mtDNA (which can be as much as 30% divergent) in germline cells. Paternally inherited mitochondria have additionally been reported in some insects such as fruit flies,honeybees, and periodical cicadas.
Mitochondrial donation
An IVF technique known as mitochondrial donation or mitochondrial replacement therapy (MRT) results in offspring containing mtDNA from a donor female, and nuclear DNA from the mother and father. In the spindle transfer procedure, the nucleus of an egg is inserted into the cytoplasm of an egg from a donor female which has had its nucleus removed, but still contains the donor female's mtDNA. The composite egg is then fertilized with the male's sperm. The procedure is used when a woman with genetically defective mitochondria wishes to procreate and produce offspring with healthy mitochondria. The first known child to be born as a result of mitochondrial donation was a boy born to a Jordanian couple in Mexico on 6 April 2016.
Mutations and disease
Susceptibility
The concept that mtDNA is particularly susceptible to reactive oxygen species generated by the respiratory chain due to its proximity remains controversial. mtDNA does not accumulate any more oxidative base damage than nuclear DNA. It has been reported that at least some types of oxidative DNA damage are repaired more efficiently in mitochondria than they are in the nucleus. mtDNA is packaged with proteins which appear to be as protective as proteins of the nuclear chromatin. Moreover, mitochondria evolved a unique mechanism which maintains mtDNA integrity through degradation of excessively damaged genomes followed by replication of intact/repaired mtDNA. This mechanism is not present in the nucleus and is enabled by multiple copies of mtDNA present in mitochondria. The outcome of mutation in mtDNA may be an alteration in the coding instructions for some proteins, which may have an effect on organism metabolism and/or fitness.
Genetic illness
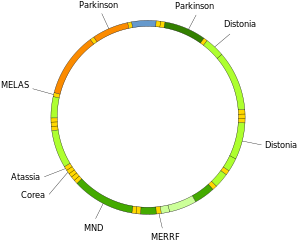
Mutations of mitochondrial DNA can lead to a number of illnesses including exercise intolerance and Kearns–Sayre syndrome (KSS), which causes a person to lose full function of heart, eye, and muscle movements. Some evidence suggests that they might be major contributors to the aging process and age-associated pathologies. Particularly in the context of disease, the proportion of mutant mtDNA molecules in a cell is termed heteroplasmy. The within-cell and between-cell distributions of heteroplasmy dictate the onset and severity of disease and are influenced by complicated stochastic processes within the cell and during development.
Mutations in mitochondrial tRNAs can be responsible for severe diseases like the MELAS and MERRF syndromes.
Mutations in nuclear genes that encode proteins that mitochondria use can also contribute to mitochondrial diseases. These diseases do not follow mitochondrial inheritance patterns, but instead follow Mendelian inheritance patterns.
Use in disease diagnosis
Recently a mutation in mtDNA has been used to help diagnose prostate cancer in patients with negative prostate biopsy. mtDNA alterations can be detected in the bio-fluids of patients with cancer. mtDNA is characterized by the high rate of polymorphisms and mutations. Some of which are increasingly recognized as an important cause of human pathology such as oxidative phosphorylation (OXPHOS) disorders, maternally inherited diabetes and deafness (MIDD), Type 2 diabetes mellitus, Neurodegenerative disease, heart failure and cancer.
Relationship with ageing
Though the idea is controversial, some evidence suggests a link between aging and mitochondrial genome dysfunction. In essence, mutations in mtDNA upset a careful balance of reactive oxygen species (ROS) production and enzymatic ROS scavenging (by enzymes like superoxide dismutase, catalase, glutathione peroxidase and others). However, some mutations that increase ROS production (e.g., by reducing antioxidant defenses) in worms increase, rather than decrease, their longevity. Also, naked mole rats, rodents about the size of mice, live about eight times longer than mice despite having reduced, compared to mice, antioxidant defenses and increased oxidative damage to biomolecules. Once, there was thought to be a positive feedback loop at work (a 'Vicious Cycle'); as mitochondrial DNA accumulates genetic damage caused by free radicals, the mitochondria lose function and leak free radicals into the cytosol. A decrease in mitochondrial function reduces overall metabolic efficiency. However, this concept was conclusively disproved when it was demonstrated that mice, which were genetically altered to accumulate mtDNA mutations at accelerated rate do age prematurely, but their tissues do not produce more ROS as predicted by the 'Vicious Cycle' hypothesis. Supporting a link between longevity and mitochondrial DNA, some studies have found correlations between biochemical properties of the mitochondrial DNA and the longevity of species. The application of a mitochondrial-specific ROS scavenger, which lead to a significant longevity of the mice studied, suggests that mitochondria may still be well-implicated in ageing. Extensive research is being conducted to further investigate this link and methods to combat ageing. Presently, gene therapy and nutraceutical supplementation are popular areas of ongoing research. Bjelakovic et al. analyzed the results of 78 studies between 1977 and 2012, involving a total of 296,707 participants, and concluded that antioxidant supplements do not reduce all-cause mortality nor extend lifespan, while some of them, such as beta carotene, vitamin E, and higher doses of vitamin A, may actually increase mortality. In a recent study, it was showed that dietary restriction can reverse ageing alterations by affecting the accumulation of mtDNA damage in several organs of rats. For example, dietary restriction prevented age-related accumulation of mtDNA damage in the cortex and decreased it in the lung and testis.
Neurodegenerative diseases
Increased mtDNA damage is a feature of several neurodegenerative diseases.
The brains of individuals with Alzheimer’s disease have elevated levels of oxidative DNA damage in both nuclear DNA and mtDNA, but the mtDNA has approximately 10-fold higher levels than nuclear DNA. It has been proposed that aged mitochondria is the critical factor in the origin of neurodegeneration in Alzheimer’s disease. Analysis of the brains of AD patients suggested an impaired function of the DNA repair pathway, which would cause reduce the overall quality of mtDNA.
In Huntington’s disease, mutant huntingtin protein causes mitochondrial dysfunction involving inhibition of mitochondrial electron transport, higher levels of reactive oxygen species and increased oxidative stress. Mutant huntingtin protein promotes oxidative damage to mtDNA, as well as nuclear DNA, that may contribute to Huntington’s disease pathology.
The DNA oxidation product 8-oxoguanine (8-oxoG) is a well-established marker of oxidative DNA damage. In persons with amyotrophic lateral sclerosis (ALS), the enzymes that normally repair 8-oxoG DNA damages in the mtDNA of spinal motor neurons are impaired. Thus oxidative damage to mtDNA of motor neurons may be a significant factor in the etiology of ALS.
Correlation of the mtDNA base composition with animal life spans
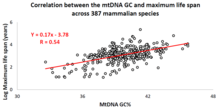
Over the past decade, an Israeli research group led by Professor Vadim Fraifeld has shown that strong and significant correlations exist between the mtDNA base composition and animal species-specific maximum life spans. As demonstrated in their work, higher mtDNA guanine + cytosine content (GC%) strongly associates with longer maximum life spans across animal species. An additional observation is that the mtDNA GC% correlation with the maximum life spans is independent of the well-known correlation between animal species metabolic rate and maximum life spans. The mtDNA GC% and resting metabolic rate explain the differences in animal species maximum life spans in a multiplicative manner (i.e., species maximum life span = their mtDNA GC% * metabolic rate). To support the scientific community in carrying out comparative analyses between mtDNA features and longevity across animals, a dedicated database was built named MitoAge.
mtDNA mutational spectrum is sensitive to species-specific life-history traits
De novo mutations arise either due to mistakes during DNA replication or due to unrepaired damage caused in turn by endogenous and exogenous mutagens. It has been long believed that mtDNA can be particularly sensitive to damage caused by reactive oxygen species (ROS), however G>T substitutions, the hallmark of the oxidative damage in the nuclear genome, are very rare in mtDNA and do not increase with age. Comparing the mtDNA mutational spectra of hundreds of mammalian species, it has been recently demonstrated that species with extended lifespans have an increased rate of A>G substitutions on single-stranded heavy chain. This discovery led to the hypothesis that A>G is a mitochondria-specific marker of age-associated oxidative damage. This finding provides a mutational (contrary to the selective one) explanation for the observation that long-lived species have GC-rich mtDNA: long-lived species become GC-rich simply because of their biased process of mutagenesis. An association between mtDNA mutational spectrum and species-specific life-history traits in mammals opens a possibility to link these factors together discovering new life-history-specific mutagens in different groups of organisms.
Relationship with non-B (non-canonical) DNA structures
Deletion breakpoints frequently occur within or near regions showing non-canonical (non-B) conformations, namely hairpins, cruciforms and cloverleaf-like elements. Moreover, there is data supporting the involvement of helix-distorting intrinsically curved regions and long G-tetrads in eliciting instability events. In addition, higher breakpoint densities were consistently observed within GC-skewed regions and in the close vicinity of the degenerate sequence motif YMMYMNNMMHM.
Use in forensics
Unlike nuclear DNA, which is inherited from both parents and in which genes are rearranged in the process of recombination, there is usually no change in mtDNA from parent to offspring. Although mtDNA also recombines, it does so with copies of itself within the same mitochondrion. Because of this and because the mutation rate of animal mtDNA is higher than that of nuclear DNA, mtDNA is a powerful tool for tracking ancestry through females (matrilineage) and has been used in this role to track the ancestry of many species back hundreds of generations.
mtDNA testing can be used by forensic scientists in cases where nuclear DNA is severely degraded. Autosomal cells only have two copies of nuclear DNA, but can have hundreds of copies of mtDNA due to the multiple mitochondria present in each cell. This means highly degraded evidence that would not be beneficial for STR analysis could be used in mtDNA analysis. mtDNA may be present in bones, teeth, or hair, which could be the only remains left in the case of severe degradation. In contrast to STR analysis, mtDNA sequencing uses Sanger sequencing. The known sequence and questioned sequence are both compared to the Revised Cambridge Reference Sequence to generate their respective haplotypes. If the known sample sequence and questioned sequence originated from the same matriline, one would expect to see identical sequences and identical differences from the rCRS. Cases arise where there are no known samples to collect and the unknown sequence can be searched in a database such as EMPOP. The Scientific Working Group on DNA Analysis Methods recommends three conclusions for describing the differences between a known mtDNA sequence and a questioned mtDNA sequence: exclusion for two or more differences between the sequences, inconclusive if there is one nucleotide difference, or cannot exclude if there are no nucleotide differences between the two sequences.
The rapid mutation rate (in animals) makes mtDNA useful for assessing genetic relationships of individuals or groups within a species and also for identifying and quantifying the phylogeny (evolutionary relationships; see phylogenetics) among different species. To do this, biologists determine and then compare the mtDNA sequences from different individuals or species. Data from the comparisons is used to construct a network of relationships among the sequences, which provides an estimate of the relationships among the individuals or species from which the mtDNAs were taken. mtDNA can be used to estimate the relationship between both closely related and distantly related species. Due to the high mutation rate of mtDNA in animals, the 3rd positions of the codons change relatively rapidly, and thus provide information about the genetic distances among closely related individuals or species. On the other hand, the substitution rate of mt-proteins is very low, thus amino acid changes accumulate slowly (with corresponding slow changes at 1st and 2nd codon positions) and thus they provide information about the genetic distances of distantly related species. Statistical models that treat substitution rates among codon positions separately, can thus be used to simultaneously estimate phylogenies that contain both closely and distantly related species
Mitochondrial DNA was admitted into evidence for the first time ever in a United States courtroom in 1996 during State of Tennessee v. Paul Ware.
In the 1998 United States court case of Commonwealth of Pennsylvania v. Patricia Lynne Rorrer, mitochondrial DNA was admitted into evidence in the State of Pennsylvania for the first time. The case was featured in episode 55 of season 5 of the true crime drama series Forensic Files (season 5).
Mitochondrial DNA was first admitted into evidence in California, United States, in the successful prosecution of David Westerfield for the 2002 kidnapping and murder of 7-year-old Danielle van Dam in San Diego: it was used for both human and dog identification. This was the first trial in the U.S. to admit canine DNA.
The remains of King Richard III, who died in 1485, were identified by comparing his mtDNA with that of two matrilineal descendants of his sister who were alive in 2013, 527 years after he died.
Use in evolutionary biology and systematic biology
mtDNA is conserved across eukaryotic organism given the critical role of mitochondria in cellular respiration. However, due to less efficient DNA repair (compared to nuclear DNA) it has a relatively high mutation rate (but slow compared to other DNA regions such as microsatellites) which makes it useful for studying the evolutionary relationships—phylogeny—of organisms. Biologists can determine and then compare mtDNA sequences among different species and use the comparisons to build an evolutionary tree for the species examined.
For instance, while most nuclear genes are nearly identical between humans and chimpanzees, their mitochondrial genomes are 9.8% different. Human and gorilla mitochondrial genomes are 11.8% different, suggesting that humans may be more closely related to chimpanzees than gorillas.
mtDNA in nuclear DNA
Whole genome sequences of more than 66,000 people revealed that most of them had some mitochondrial DNA inserted into their nuclear genomes. More than 90% of these nuclear-mitochondrial segments (NUMTs) were inserted after humans diverged from apes. Results indicate such transfers currently occur as frequent as once in every ~4,000 human births.
It appears that organellar DNA is much more often transferred to nuclear DNA than previously thought. This observation also supports the idea of the endosymbiont theory that eukaryotes have evolved from endosymbionts which turned into organelles while transferring most of their DNA to the nucleus so that the organellar genome shrunk in the process.
History
Mitochondrial DNA was discovered in the 1960s by Margit M. K. Nass and Sylvan Nass by electron microscopy as DNase-sensitive threads inside mitochondria, and by Ellen Haslbrunner, Hans Tuppy and Gottfried Schatz by biochemical assays on highly purified mitochondrial fractions.
Mitochondrial sequence databases
Several specialized databases have been founded to collect mitochondrial genome sequences and other information. Although most of them focus on sequence data, some of them include phylogenetic or functional information.
- AmtDB: a database of ancient human mitochondrial genomes.
- InterMitoBase: an annotated database and analysis platform of protein-protein interactions for human mitochondria. (apparently last updated in 2010, but still available)
- MitoBreak: the mitochondrial DNA breakpoints database.
- MitoFish and MitoAnnotator: a mitochondrial genome database of fish. See also Cawthorn et al.
- Mitome: a database for comparative mitochondrial genomics in metazoan animals (no longer available)
- MitoRes: a resource of nuclear-encoded mitochondrial genes and their products in metazoa (apparently no longer being updated)
- MitoSatPlant: Mitochondrial microsatellites database of viridiplantae.
- MitoZoa 2.0: a database for comparative and evolutionary analyses of mitochondrial genomes in Metazoa. (no longer available)
MtDNA-phenotype association databases
Genome-wide association studies can reveal associations of mtDNA genes and their mutations with phenotypes including lifespan and disease risks. In 2021, the largest, UK Biobank-based, genome-wide association study of mitochondrial DNA unveiled 260 new associations with phenotypes including lifespan and disease risks for e.g. type 2 diabetes.
Mitochondrial mutation databases
Several specialized databases exist that report polymorphisms and mutations in the human mitochondrial DNA, together with the assessment of their pathogenicity.
- MitImpact: A collection of pre-computed pathogenicity predictions for all nucleotide changes that cause non-synonymous substitutions in human mitochondrial protein coding genes MitImpact 3D - IRCCS-CSS Bioinformatics lab.
- MITOMAP: A compendium of polymorphisms and mutations in human mitochondrial DNA WebHome < MITOMAP < Foswiki.
See also
External links
-
 Media related to Mitochondrial DNA at Wikimedia Commons
Media related to Mitochondrial DNA at Wikimedia Commons
|
Types of nucleic acids
| |||||||
|---|---|---|---|---|---|---|---|
| Constituents | |||||||
|
Ribonucleic acids |
|
||||||
| Deoxyribonucleic acids |
|||||||
| Analogues | |||||||
| Cloning vectors | |||||||