
| Sickle cell disease | |
|---|---|
| Other names | Sickle cell disorder |
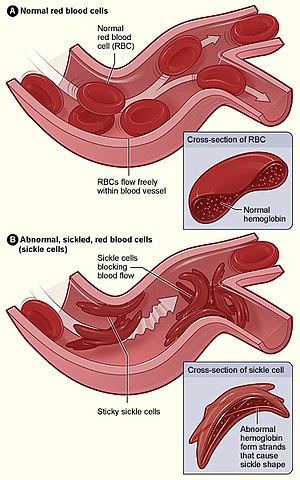 | |
| Figure (A) shows normal red blood cells flowing freely through a blood vessel. The inset shows a cross-section of a normal red blood cell with normal haemoglobin. Figure (B) shows abnormal, sickled red blood cells sticking at the branching point in a blood vessel. The inset image shows a cross-section of a sickle cell with long polymerized sickle haemoglobin (HbS) strands stretching and distorting the cell shape to look like a crescent moon. | |
| Specialty | Hematology, medical genetics |
| Symptoms | Attacks of pain, anemia, swelling in the hands and feet, bacterial infections, stroke |
| Complications | Chronic pain, stroke, aseptic bone necrosis, gallstones, leg ulcers, priapism, pulmonary hypertension, vision problems, kidney problems |
| Usual onset | 5–6 months of age |
| Causes | Genetic, Homozygous mutation in the hemoglobin S gene. |
| Diagnostic method | Blood test |
| Treatment | Vaccination, antibiotics, high fluid intake, folic acid supplementation, pain medication, blood transfusions |
| Prognosis | Life expectancy 40–60 years (developed world) |
| Frequency | 4.4 million (2015) |
| Deaths | 114,800 (2015) |
Sickle cell disease (SCD) is a group of blood disorders typically inherited. The most common type is known as sickle cell anaemia. It results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells. This leads to a rigid, sickle-like shape under certain circumstances. Problems in sickle cell disease typically begin around 5 to 6 months of age. A number of health problems may develop, such as attacks of pain (known as a sickle cell crisis), anemia, swelling in the hands and feet, bacterial infections, and stroke.Long-term pain may develop as people get older. The average life expectancy in the developed world is 40 to 60 years.
Sickle cell disease occurs when a person inherits two abnormal copies of the β-globin gene (HBB) that makes haemoglobin, one from each parent. This gene occurs in chromosome 11. Several subtypes exist, depending on the exact mutation in each haemoglobin gene. An attack can be set off by temperature changes, stress, dehydration, and high altitude. A person with a single abnormal copy does not usually have symptoms and is said to have sickle cell trait. Such people are also referred to as carriers. Diagnosis is by a blood test, and some countries test all babies at birth for the disease. Diagnosis is also possible during pregnancy.
The care of people with sickle cell disease may include infection prevention with vaccination and antibiotics, high fluid intake, folic acid supplementation, and pain medication. Other measures may include blood transfusion and the medication hydroxycarbamide (hydroxyurea). A small percentage of people can be cured by a transplant of bone marrow cells.
As of 2015, about 4.4 million people have sickle cell disease, while an additional 43 million have sickle cell trait. About 80% of sickle cell disease cases are believed to occur in Sub-Saharan Africa. It also occurs to a lesser degree in parts of India, Southern Europe, West Asia, North Africa and among people of African origin (sub-Saharan) living in other parts of the world. In 2015, it resulted in about 114,800 deaths. The condition was first described in the medical literature by American physician James B. Herrick in 1910. In 1949, its genetic transmission was determined by E. A. Beet and J. V. Neel. In 1954, the protective effect against malaria of sickle cell trait was described.
Signs and symptoms
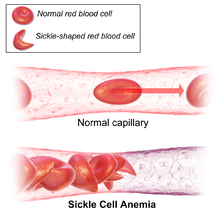
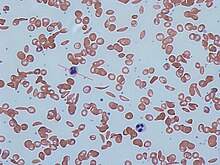

Signs of sickle cell disease usually begin in early childhood. The severity of symptoms can vary from person to person. Sickle cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.
Sickle cell crisis
The terms "sickle cell crisis" or "sickling crisis" may be used to describe several independent acute conditions occurring in patients with SCD, which results in anaemia and crises that could be of many types, including the vaso-occlusive crisis, aplastic crisis, splenic sequestration crisis, haemolytic crisis, and others. Most episodes of sickle cell crises last between five and seven days. "Although infection, dehydration, and acidosis (all of which favor sickling) can act as triggers, in most instances, no predisposing cause is identified."
Vaso-occlusive crisis
The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis, and often organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration, analgesics, and blood transfusion; pain management requires opioid drug administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manages on nonsteroidal anti-inflammatory drugs such as diclofenac or naproxen. For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia devices are commonly used in this setting. Vaso-occlusive crisis involving organs such as the penis or lungs are considered an emergency and treated with red blood cell transfusions. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.
Splenic sequestration crisis
The spleen is frequently affected in sickle cell disease, as the sickle-shaped red blood cells cause narrowing of blood vessels and reduced function in clearing the defective cells. It is usually infarcted before the end of childhood in individuals with sickle cell anaemia. This spleen damage increases the risk of infection from encapsulated organisms; preventive antibiotics and vaccinations are recommended for those lacking proper spleen function.
Splenic sequestration crises are acute, painful enlargements of the spleen, caused by intrasplenic trapping of red cells and resulting in a precipitous fall in haemoglobin levels with the potential for hypovolemic shock. Sequestration crises are considered an emergency. If not treated, patients may die within 1–2 hours due to circulatory failure. Management is supportive, sometimes with blood transfusion. These crises are transient; they continue for 3–4 hours and may last for one day.
Acute chest syndrome
Acute chest syndrome is defined by at least two of these signs or symptoms: chest pain, fever, pulmonary infiltrate or focal abnormality, respiratory symptoms, or hypoxemia. It is the second-most common complication and it accounts for about 25% of deaths in patients with SCD. Most cases present with vaso-occlusive crises, and then develop acute chest syndrome. Nevertheless, about 80% of people have vaso-occlusive crises during acute chest syndrome.
Aplastic crisis
Aplastic crises are instances of an acute worsening of the patient's baseline anaemia, producing pale appearance, fast heart rate, and fatigue. This crisis is normally triggered by parvovirus B19, which directly affects production of red blood cells by invading the red cell precursors and multiplying in and destroying them. Parvovirus infection almost completely prevents red blood cell production for two to three days. In normal individuals, this is of little consequence, but the shortened red cell life of SCD patients results in an abrupt, life-threatening situation. Reticulocyte counts drop dramatically during the disease (causing reticulocytopenia), and the rapid turnover of red cells leads to the drop in haemoglobin. This crisis takes 4 to 7 days to disappear. Most patients can be managed supportively; some need a blood transfusion.
Haemolytic crisis
Haemolytic crises are acute accelerated drops in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in people with coexistent G6PD deficiency. Another influence of hemolytic crises in sickle cell disease is oxidative stress on the erythrocytes, leukocytes, and platelets. When there is not enough red blood cell production in the bone marrow, the oxygen that the body receives, processes, and transports is unbalanced with the body's antioxidants. There is an imbalance in the oxygen reactive species in the cells, which leads to more production of red blood cells that are not properly oxygenated or formed. Oxidative stress may lead to anemia because of the imbalance of oxygen in the tissue.
Management is supportive, sometimes with blood transfusions.
Other
One of the earliest clinical manifestations is dactylitis, presenting as early as six months of age, and may occur in children with sickle cell trait. The crisis can last up to a month. Given that pneumonia and sickling in the lung can both produce symptoms of acute chest syndrome, the patient is treated for both conditions. It can be triggered by painful crisis, respiratory infection, bone-marrow embolisation, or possibly by atelectasis, opiate administration, or surgery.Hematopoietic ulcers may also occur.
Complications
Sickle cell anaemia can lead to various complications, including:
- Increased risk of severe bacterial infections is due to loss of functioning spleen tissue (and comparable to the risk of infections after having the spleen removed surgically). These infections are typically caused by encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae. Daily penicillin prophylaxis is the most commonly used treatment during childhood, with some haematologists continuing treatment indefinitely. Patients benefit today from routine vaccination for S. pneumoniae.
- Stroke, which can result from a progressive narrowing of blood vessels, prevents oxygen from reaching the brain. Cerebral infarction occurs in children and cerebral haemorrhage in adults.
- Silent stroke causes no immediate symptoms, but is associated with damage to the brain. Silent stroke is probably five times as common as symptomatic stroke. About 10–15% of children with SCD have strokes, with silent strokes predominating in the younger patients.
- Cholelithiasis (gallstones) and cholecystitis may result from excessive bilirubin production and precipitation due to prolonged haemolysis.
- Avascular necrosis (aseptic bone necrosis) of the hip and other major joints may occur as a result of ischaemia.
- Decreased immune reactions due to hyposplenism (malfunctioning of the spleen)
- Priapism and infarction of the penis
- Osteomyelitis (bacterial bone infection), the most common cause of osteomyelitis in SCD is Salmonella (especially the atypical serotypes Salmonella typhimurium, Salmonella enteritidis, Salmonella choleraesuis, and Salmonella paratyphi B), followed by Staphylococcus aureus and Gram-negative enteric bacilli perhaps because intravascular sickling of the bowel leads to patchy ischaemic infarction.
- Acute papillary necrosis in the kidneys
- Leg ulcers
- In eyes, background retinopathy, proliferative retinopathy, vitreous haemorrhages, and retinal detachments can result in blindness. Regular annual eye checks are recommended.
- During pregnancy, intrauterine growth restriction, spontaneous abortion, and pre-eclampsia
- Chronic pain: Even in the absence of acute vaso-occlusive pain, many patients have unreported chronic pain.
- Pulmonary hypertension (increased pressure on the pulmonary artery) can lead to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance, and episodes of syncope. 21% of children and 30% of adults have evidence of pulmonary hypertension when tested; this is associated with reduced walking distance and increased mortality.
- Cardiomyopathy and left ventricular diastolic dysfunction caused by fibrosis or scarring of cardiac tissues. This also contributes to pulmonary hypertension, decreased exercise capacity, and arrhythmias.
- Chronic kidney failure due to sickle-cell nephropathy manifests itself with hypertension, protein loss in the urine, loss of red blood cells in urine and worsened anaemia. If it progresses to end-stage kidney failure, it carries a poor prognosis.
Genetics


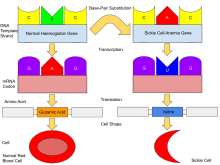
Normally, humans have haemoglobin A, which consists of two alpha and two beta chains, haemoglobin A2, which consists of two alpha and two delta chains, and haemoglobin F (HbF), consisting of two alpha and two gamma chains in their bodies. Of these three types, haemoglobin F dominates until about 6 weeks of age. Afterwards, haemoglobin A dominates throughout life. In people diagnosed with sickle cell disease, at least one of the β-globin subunits in haemoglobin A is replaced with what is known as haemoglobin S. In sickle cell anaemia, a common form of sickle cell disease, haemoglobin S replaces both β-globin subunits in the haemoglobin.
Sickle cell disease has an autosomal recessive pattern of inheritance from parents. The types of haemoglobin a person makes in the red blood cells depend on what haemoglobin genes are inherited from her or his parents. If one parent has sickle cell anaemia and the other has sickle cell trait, then the child has a 50% chance of having sickle cell disease and a 50% chance of having sickle cell trait. When both parents have sickle cell trait, a child has a 25% chance of sickle cell disease; 25% do not carry any sickle cell alleles, and 50% have the heterozygous condition.
Sickle cell gene mutation probably arose spontaneously in different geographic areas, as suggested by restriction endonuclease analysis. These variants are known as Cameroon, Senegal, Benin, Bantu, and Saudi-Asian. Their clinical importance is because some are associated with higher HbF levels, e.g., Senegal and Saudi-Asian variants, and tend to have milder disease.
The gene defect is a single nucleotide mutation (see single-nucleotide polymorphism – SNP) (GAG codon changing to GTG) of the β-globin gene, which results in glutamate (E/Glu) being substituted by valine (V/Val) at position 6 (E6V substitution). Haemoglobin S with this mutation is referred to as HbS, as opposed to the normal adult HbA. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structures of haemoglobin in conditions of normal oxygen concentration. However, under low oxygen concentration, HbS polymerizes and forms fibrous precipitates because the deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices (Phe 85, Leu 88).
In people heterozygous for HbS (carriers of sickling haemoglobin), the polymerisation problems are minor because the normal allele can produce half of the haemoglobin. In people homozygous for HbS, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smooth, doughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated.
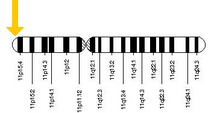
The allele responsible for sickle cell anaemia can be found on the short arm of chromosome 11, more specifically 11p15.5. A person who receives the defective gene from both father and mother develops the disease; a person who receives one defective and one healthy allele remains healthy, but can pass on the disease and is known as a carrier or heterozygote. Heterozygotes are still able to contract malaria, but their symptoms are generally less severe.
Due to the adaptive advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India, and the Middle East. Malaria was historically endemic to southern Europe, but it was declared eradicated in the mid-20th century, with the exception of rare sporadic cases.
The malaria parasite has a complex lifecycle and spends part of it in red blood cells. In a carrier, the presence of the malaria parasite causes the red blood cells with defective haemoglobin to rupture prematurely, making the Plasmodium parasite unable to reproduce. Further, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people's chances of survival actually increase if they carry sickle cell traits (selection for the heterozygote).
In the United States, with no endemic malaria, the prevalence of sickle cell anaemia among people of African ancestry is lower (about 0.25%) than among people in West Africa (about 4.0%) and is falling. Without endemic malaria, the sickle cell mutation is purely disadvantageous and tends to decline in the affected population by natural selection, and now artificially through prenatal genetic screening. However, the African American community descends from a significant admixture of several African and non-African ethnic groups and also represents the descendants of survivors of slavery and the slave trade. Thus, a degree of genetic dilution via crossbreeding with non-African people and high health-selective pressure through slavery (especially the slave trade and the frequently deadly Middle Passage) may be the most plausible explanations for the lower prevalence of sickle cell anaemia (and, possibly, other genetic diseases) among African Americans compared to West Africans. Another factor that limits the spread of sickle cell genes in North America is the relative absence of polygamy. In polygamous societies, affected males may father many children with multiple partners.
Pathophysiology
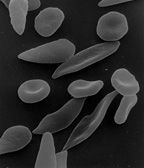
The loss of red blood cell elasticity is central to the pathophysiology of sickle cell disease. Normal red blood cells are quite elastic and have a biconcave disc shape, which allows the cells to deform to pass through capillaries. In sickle cell disease, low oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell's elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells, because of their shape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction. Healthy red blood cells typically function for 90–120 days, but sickled cells only last 10–20 days.
Diagnosis
In HbS, the complete blood count reveals haemoglobin levels in the range of 6–8 g/dl with a high reticulocyte count (as the bone marrow compensates for the destruction of sickled cells by producing more red blood cells). In other forms of sickle cell disease, Hb levels tend to be higher. A blood film may show features of hyposplenism (target cells and Howell-Jolly bodies).
Sickling of the red blood cells, on a blood film, can be induced by the addition of sodium metabisulfite. The presence of sickle haemoglobin can also be demonstrated with the "sickle solubility test" (also called "sickledex"). A mixture of haemoglobin S (HbS) in a reducing solution (such as sodium dithionite) gives a turbid appearance, whereas normal Hb gives a clear solution.
Abnormal haemoglobin forms can be detected on haemoglobin electrophoresis, a form of gel electrophoresis on which the various types of haemoglobin move at varying speeds. Sickle cell haemoglobin (HgbS) and haemoglobin C with sickling (HgbSC)—the two most common forms—can be identified from there. The diagnosis can be confirmed with high-performance liquid chromatography. Genetic testing is rarely performed, as other investigations are highly specific for HbS and HbC.
An acute sickle cell crisis is often precipitated by infection. Therefore, a urinalysis to detect an occult urinary tract infection, and chest X-ray to look for occult pneumonia should be routinely performed.
People who are known carriers of the disease or at risk of having a child with sickle cell anemia may undergo genetic counseling. Genetic counselors work with families to discuss the benefits, limitations, and logistics of genetic testing options as well as the potential impact of testing and test results on the individual. During pregnancy, genetic testing can be done on either a blood sample from the fetus or a sample of amniotic fluid. During the first trimester of pregnancy, chorionic villus sampling (CVS) is also a technique used for SCD prenatal diagnosis. Since taking a blood sample from a fetus has greater risks, the latter test is usually used. Neonatal screening sometimes referred to as newborn screening, provides not only a method of early detection for individuals with sickle cell disease but also allows for the identification of the groups of people who carry the sickle cell trait. Genetic counselors can help individuals of colour and their families tackle the racial and ethnic disparities that exist in healthcare.
In 2010, there was significant consideration and debate in the US surrounding comprehensive screening of athletes for SCD. The American Society of Hematology concluded in a statement in 2012 that they do not support testing or disclosure of sickle cell trait status as a prerequisite for participation in athletic activities due to lack of scientific evidence, inconsistency with good medical practice, and inconsistency with public health ethics. They recommended universal interventions to reduce exertion-related injuries and deaths effective for all athletes irrespective of their sickle cell status.
Management
Treatment involves a number of measures. While it has been historically recommended that people with sickle cell disease avoid exercise, regular exercise may benefit people. Dehydration should be avoided. A diet high in calcium is recommended but the effectiveness of vitamin D supplementation remains uncertain.L-glutamine use was supported by the FDA starting at the age of five, as it decreases complications.
Folic acid and penicillin
From birth to five years of age, penicillin daily, due to the immature immune system that makes them more prone to early childhood illnesses, is recommended. Dietary supplementation of folic acid had been previously recommended by the WHO. A 2016 Cochrane review of its use found "the effect of supplementation on anaemia and any symptoms of anaemia remains unclear" due to a lack of medical evidence.
Malaria prevention

The protective effect of sickle cell trait does not apply to people with sickle cell disease; in fact, they are more vulnerable to malaria, since the most common cause of painful crises in malarial countries is infection with malaria. People with sickle cell disease living in malarial countries should receive lifelong medication for prevention.
Vaso-occlusive crisis
Most people with sickle cell disease have intensely painful episodes called vaso-occlusive crises. However, the frequency, severity, and duration of these crises vary tremendously. Painful crises are treated symptomatically with pain medications; pain management requires opioid drug administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manages on NSAIDs (such as diclofenac or naproxen). For more severe crises, most patients require inpatient management for intravenous opioids.
Extra fluids, administered either orally or intravenously, are a routine part of treatment of vaso-occlusive crises but the evidence about the most effective route, amount and type of fluid replacement remains uncertain.
Crizanlizumab, a monoclonal antibody target towards p-selectin was approved in 2019 in the United States to reduce the frequency of vaso-occlusive crisis in those 16 years and older.
Stroke prevention
Transcranial Doppler ultrasound (TCD) can detect children with sickle cell that have a high risk for stroke. The ultrasound test detects blood vessels partially obstructed by sickle cells by measuring the rate of blood into the brain, as blood flow velocity is inversely related to arterial diameter, and consequently, high blood-flow velocity is correlated with narrowing of the arteries. In 2002 the National Institute of Health (NIH) issued a statement recommending that children with sickle cell get the Transcranial Doppler ultrasound screen annually, and in 2014 a panel of experts convened by the NIH issued guidelines reiterating the same recommendation. One review of medical records, by hematologist Dr. Julie Kanter at the University of Alabama at Birmingham, showed that on average only 48.4 percent of children with sickle cell get the recommended ultrasound test.
A 1994 NIH study showed that children at risk for strokes who received blood transfusions had an annual stroke rate of less than 1 percent, whereas those children who did not receive blood transfusions had a 10 percent stroke rate per year. (Also see 1998 study in the New England Journal of Medicine.) In addition to ultrasounds and blood transfusions, the inexpensive generic drug hydroxyurea can reduce the risk of irreversible organ and brain damage. Guidelines from NIH published in 2014 state that all children and adolescents should take hydroxyurea, as should adults with serious complications or three or more pain crises in a year.
Acute chest syndrome
Management is similar to vaso-occlusive crisis, with the addition of antibiotics (usually a quinolone or macrolide, since cell wall-deficient ["atypical"] bacteria are thought to contribute to the syndrome), oxygen supplementation for hypoxia, and close observation. In the absence of high quality evidence regarding the effectiveness of antibiotics for acute chest syndrome in people with sickle cell disease, there is no standard antibiotic treatment as of 2019. It is recommended that people with suspected acute chest syndrome should be admitted to the hospital with worsening A-a gradient an indication for ICU admission.
Should the pulmonary infiltrate worsen or the oxygen requirements increase, simple blood transfusion or exchange transfusion is indicated. The latter involves the exchange of a significant portion of the person's red cell mass for normal red cells, which decreases the level of haemoglobin S in the patient's blood. However, there is currently uncertain evidence about the possible benefits or harms of blood transfusion for acute chest syndrome in people with sickle cell disease.
Hydroxyurea
Hydroxyurea, also known as hydroxycarbamide, probably reduces the frequency of painful episodes and the risk of life-threatening illness or death but there is currently insufficient evidence regarding the risk of adverse effects. Hydroxyurea and phlebotomy combined may be more effective than transfusion and chelation combined in terms of pain, life-threatening illness and risk of death.
It was the first approved drug for the treatment of sickle cell anaemia, and was shown to decrease the number and severity of attacks in 1995 and shown to possibly increase survival time in a study in 2003. This is achieved, in part, by reactivating fetal haemoglobin production in place of the haemoglobin S that causes sickle cell anaemia. Hydroxyurea had previously been used as a chemotherapy agent, and some concern exists that long-term use may be harmful, but this risk is either absent or very small and the benefits likely outweigh the risks.
Voxelotor was approved in the United States in 2019 to increase hemoglobin in people with SS disease.
Blood transfusion
Blood transfusions are often used in the management of sickle cell disease in acute cases and to prevent complications by decreasing the number of red blood cells (RBCs) that can sickle by adding normal red blood cells. In children, preventive RBC transfusion therapy has been shown to reduce the risk of first stroke or silent stroke when transcranial Doppler ultrasonography shows abnormal cerebral blood flow. In those who have sustained a prior stroke event, it also reduces the risk of recurrent stroke and additional silent strokes.
Bone marrow transplant
Bone marrow transplants have proven effective in children; they are the only known cure for SCD. However, bone marrow transplants are difficult to obtain because of the specific HLA typing necessary. Ideally, a close relative (allogeneic) would donate the bone marrow necessary for transplantation. Some gene therapies are under development that would alter the patient's own bone marrow stem cells ex vivo, which can then be transplanted back into the patient after chemotherapy eliminates the original unmodified cells.
Avascular necrosis
When treating avascular necrosis of the bone in people with sickle cell disease, the aim of treatment is to reduce or stop the pain and maintain joint mobility. Current treatment options include resting the joint, physical therapy, pain-relief medicine, joint-replacement surgery, or bone grafting. High quality, randomized, controlled trials are needed to assess the most effective treatment option and determine if a combination of physical therapy and surgery is more effective than physical therapy alone.
Psychological therapies
Psychological therapies such as patient education, cognitive therapy, behavioural therapy, and psychodynamic psychotherapy, that aim to complement current medical treatments, require further research to determine their effectiveness.
Prognosis
About 90% of people survive to age 20, and close to 50% survive beyond age 50. In 2001, according to one study performed in Jamaica, the estimated mean survival for people was 53 years for men and 58 years for women with homozygous SCD. The specific life expectancy in much of the developing world is unknown. In 1975 about 7.3% of people with SCD died before their 23rd birthday; while in 1989 2.6% of people with SCD died by the age of 20.
Epidemiology
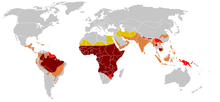
The highest frequency of sickle cell disease is found in tropical regions, particularly sub-Saharan Africa, tribal regions of India, and the Middle East. Migration of substantial populations from these high-prevalence areas to low-prevalence countries in Europe has dramatically increased in recent decades and in some European countries, sickle cell disease has now overtaken more familiar genetic conditions such as haemophilia and cystic fibrosis. In 2015, it resulted in about 114,800 deaths.
Sickle cell disease occurs more commonly among people whose ancestors lived in tropical and subtropical sub-Saharan regions where malaria is or was common. Where malaria is common, carrying a single sickle cell allele (trait) confers a heterozygote advantage; humans with one of the two alleles of sickle cell disease show less severe symptoms when infected with malaria.
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Africa
Three-quarters of sickle cell cases occur in Africa. A recent WHO report estimated that around 2% of newborns in Nigeria were affected by sickle cell anaemia, giving a total of 150,000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10 and 40% across equatorial Africa, decreasing to 1–2% on the North African coast and <1% in South Africa. Studies in Africa show a significant decrease in infant mortality rate, ages 2–16 months, because of the sickle cell trait. This happened in areas of predominant malarial cases.
Uganda has the fifth-highest sickle cell disease burden in Africa. One study indicates that 20 000 babies per year are born with sickle cell disease with the sickle cell trait at 13·3% and with disease 0·7%.
| Country | Population(2020) | Subregion | % of Prevalence | Prevalence | Incidence |
| Angola | 32,866,272 | Middle Africa | 0.09375 | 3,081,213 | 14,869 |
| Cameroon | 26,545,863 | Middle Africa | 0.117 | 3,105,866 | 11,826 |
| DR Congo | 89,561,403 | Middle Africa | 0.1163333333 | 10,418,977 | 65,536 |
| Ghana | 31,072,940 | Western Africa | 0.09375 | 2,913,088 | 9,588 |
| Guinea | 13,132,795 | Western Africa | 0.139375 | 1,830,383 | 8,907 |
| Niger | 24,206,644 | Western Africa | 0.07025 | 1,700,517 | 8,756 |
| Nigeria | 206,139,589 | Western Africa | 0.1286666667 | 26,523,294 | 150,000 |
| Tanzania | 59,734,218 | Eastern Africa | 0.0545 | 3,255,515 | 19,585 |
| Uganda | 45,741,007 | Eastern Africa | 0.07025 | 3,213,306 | 17,936 |
| Zambia | 18,383,955 | Eastern Africa | 0.082 | 1,507,484 | 9,958 |
| Algeria | 43,851,044 | Northern Africa | 0.029 | 1,271,680 | 6,624 |
| Benin | 12,123,200 | Western Africa | 0.1286666667 | 1,559,852 | 8,125 |
| Botswana | 2,351,627 | Southern Africa | 0.029 | 68,197 | 355 |
| Burkina Faso | 20,903,273 | Western Africa | 0.07025 | 1,468,455 | 7,649 |
| Burundi | 11,890,784 | Eastern Africa | 0.023 | 273,488 | 1,425 |
| Cabo Verde | 555,987 | Western Africa | 0.023 | 12,788 | 67 |
| Central African Republic | 4,829,767 | Middle Africa | 0.082 | 396,041 | 2,063 |
| Chad | 16,425,864 | Middle Africa | 0.0585 | 960,913 | 5,005 |
| Comoros | 869,601 | Eastern Africa | 0.023 | 20,001 | 104 |
| Congo | 5,518,087 | Middle Africa | 0.1615 | 891,171 | 4,642 |
| Côte d'Ivoire | 26,378,274 | Western Africa | 0.07025 | 1,853,074 | 9,652 |
| Djibouti | 988,000 | Eastern Africa | 0.023 | 22,724 | 118 |
| Egypt | 102,334,404 | Northern Africa | 0.029 | 2,967,698 | 15,458 |
| Equatorial Guinea | 1,402,985 | Middle Africa | 0.181 | 253,940 | 1,323 |
| Eritrea | 3,546,421 | Eastern Africa | 0.023 | 81,568 | 425 |
| Eswatini | 1,160,164 | Southern Africa | 0.023 | 26,684 | 139 |
| Ethiopia | 114,963,588 | Eastern Africa | 0.029 | 3,333,944 | 17,366 |
| Gabon | 2,225,734 | Middle Africa | 0.181 | 402,858 | 2,098 |
| Gambia | 2,416,668 | Western Africa | 0.082 | 198,167 | 1,032 |
| Guinea-Bissau | 1,968,001 | Western Africa | 0.035 | 68,880 | 359 |
| Kenya | 53,771,296 | Eastern Africa | 0.04675 | 2,513,808 | 13,094 |
| Lesotho | 2,142,249 | Southern Africa | 0.023 | 49,272 | 257 |
| Liberia | 5,057,681 | Western Africa | 0.07025 | 355,302 | 1,851 |
| Libya | 6,871,292 | Northern Africa | 0.029 | 199,267 | 1,038 |
| Madagascar | 27,691,018 | Eastern Africa | 0.04675 | 1,294,555 | 6,743 |
| Malawi | 19,129,952 | Eastern Africa | 0.035 | 669,548 | 3,488 |
| Mali | 20,250,833 | Western Africa | 0.082 | 1,660,568 | 8,650 |
| Mauritania | 4,649,658 | Western Africa | 0.04675 | 217,372 | 1,132 |
| Mauritius | 1,271,768 | Eastern Africa | 0.023 | 29,251 | 152 |
| Morocco | 36,910,560 | Northern Africa | 0.029 | 1,070,406 | 5,576 |
| Mozambique | 31,255,435 | Eastern Africa | 0.035 | 1,093,940 | 5,698 |
| Namibia | 2,540,905 | Southern Africa | 0.03883333333 | 98,672 | 514 |
| Rwanda | 12,952,218 | Eastern Africa | 0.035 | 453,328 | 2,361 |
| São Tomé and Príncipe | 219,159 | Middle Africa | 0.181 | 39,668 | 207 |
| Senegal | 16,743,927 | Western Africa | 0.07025 | 1,176,261 | 6,127 |
| Seychelles | 98,347 | Eastern Africa | 0.023 | 2,262 | 12 |
| Sierra Leone | 7,976,983 | Western Africa | 0.1615 | 1,288,283 | 6,711 |
| Somalia | 15,893,222 | Eastern Africa | 0.029 | 460,903 | 2,401 |
| South Africa | 59,308,690 | Southern Africa | 0.029 | 1,719,952 | 8,959 |
| South Sudan | 11,193,725 | Eastern Africa | 0.04675 | 523,307 | 2,726 |
| Sudan | 43,849,260 | Northern Africa | 0.03883333333 | 1,702,813 | 8,870 |
| Togo | 8,278,724 | Western Africa | 0.09375 | 776,130 | 4,043 |
| Tunisia | 11,818,619 | Northern Africa | 0.023 | 271,828 | 1,416 |
| Zimbabwe | 14,862,924 | Eastern Africa | 0.035 | 520,202 | 2,710 |
| Total | 1,338,826,604 | Africa | 91,868,664 | 495,726 |
United States
The number of people with the disease in the United States is about 100,000 (one in 3,300), mostly affecting Americans of sub-Saharan African descent. In the United States, about one out of 365 African-American children and one in every 16,300 Hispanic-American children have sickle cell anaemia. The life expectancy for men with SCD is approximately 42 years of age while women live approximately six years longer. An additional 2 million are carriers of the sickle cell trait. Most infants with SCD born in the United States are identified by routine neonatal screening. As of 2016 all 50 states include screening for sickle cell disease as part of their newborn screen. The newborn's blood is sampled through a heel-prick and is sent to a lab for testing. The baby must have been eating for a minimum of 24 hours before the heel-prick test can be done. Some states also require a second blood test to be done when the baby is two weeks old to ensure the results. Sickle cell anemia is the most common genetic disorder among African Americans. Approximately 8% are carriers and 1 in 375 are born with the disease. Patient advocates for sickle cell disease have complained that it gets less government and private research funding than similar rare diseases such as cystic fibrosis, with researcher Elliott Vichinsky saying this shows racial discrimination or the role of wealth in health care advocacy.
France

As a result of population growth in African-Caribbean regions of overseas France and immigration from North and sub-Saharan Africa to mainland France, sickle cell disease has become a major health problem in France. SCD has become the most common genetic disease in the country, with an overall birth prevalence of one in 2,415 in metropolitan France, ahead of phenylketonuria (one in 10,862), congenital hypothyroidism (one in 3,132), congenital adrenal hyperplasia (one in 19,008) and cystic fibrosis (one in 5,014) for the same reference period.

Since 2000, neonatal screening of SCD has been performed at the national level for all newborns defined as being "at-risk" for SCD based on ethnic origin (defined as those born to parents originating from sub-Saharan Africa, North Africa, the Mediterranean area (South Italy, Greece, and Turkey), the Arabic peninsula, the French overseas islands, and the Indian subcontinent).
United Kingdom
In the United Kingdom, between 12,000 and 15,000 people are thought to have sickle cell disease with an estimated 250,000 carriers of the condition in England alone. As the number of carriers is only estimated, all newborn babies in the UK receive a routine blood test to screen for the condition. Due to many adults in high-risk groups not knowing if they are carriers, pregnant women and both partners in a couple are offered screening so they can get counselling if they have the sickle cell trait. In addition, blood donors from those in high-risk groups are also screened to confirm whether they are carriers and whether their blood filters properly. Donors who are found to be carriers are then informed and their blood, while often used for those of the same ethnic group, is not used for those with sickle cell disease who require a blood transfusion.
West Asia
In Saudi Arabia, about 4.2% of the population carry the sickle cell trait and 0.26% have sickle cell disease. The highest prevalence is in the Eastern province, where approximately 17% of the population carry the gene and 1.2% have sickle cell disease. In 2005, Saudi Arabia introduced a mandatory premarital test including HB electrophoresis, which aimed to decrease the incidence of SCD and thalassemia.
In Bahrain, a study published in 1998 that covered about 56,000 people in hospitals in Bahrain found that 2% of newborns have sickle cell disease, 18% of the surveyed people have the sickle cell trait, and 24% were carriers of the gene mutation causing the disease. The country began screening of all pregnant women in 1992 and newborns started being tested if the mother was a carrier. In 2004, a law was passed requiring couples planning to marry to undergo free premarital counseling. These programs were accompanied by public education campaigns.
India and Nepal
Sickle cell disease is common in some ethnic groups of central India, where the prevalence has ranged from 9.4 to 22.2% in endemic areas of Madhya Pradesh, Rajasthan, and Chhattisgarh. It is also endemic among Tharu people of Nepal and India; however, they have a sevenfold lower rate of malaria despite living in a malaria infested zone.
Caribbean Islands
In Jamaica, 10% of the population carry the sickle cell gene, making it the most prevalent genetic disorder in the country.
History
The first modern report of sickle cell disease may have been in 1846, where the autopsy of an executed runaway slave was discussed; the key finding was the absence of the spleen. Reportedly, African slaves in the United States exhibited resistance to malaria, but were prone to leg ulcers. The abnormal characteristics of the red blood cells, which later lent their name to the condition, was first described by Ernest E. Irons (1877–1959), intern to Chicago cardiologist and professor of medicine James B. Herrick (1861–1954), in 1910. Irons saw "peculiar elongated and sickle-shaped" cells in the blood of a man named Walter Clement Noel, a 20-year-old first-year dental student from Grenada. Noel had been admitted to the Chicago Presbyterian Hospital in December 1904 with anaemia. Noel was readmitted several times over the next three years for "muscular rheumatism" and "bilious attacks" but completed his studies and returned to the capital of Grenada (St. George's) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada. Shortly after the report by Herrick, another case appeared in the Virginia Medical Semi-Monthly with the same title, "Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia." This article is based on a patient admitted to the University of Virginia Hospital on 15 November 1910. In the later description by Verne Mason in 1922, the name "sickle cell anemia" is first used. Childhood problems related to sickle cells disease were not reported until the 1930s, despite the fact that this cannot have been uncommon in African-American populations.
Memphis physician Lemuel Diggs, a prolific researcher into sickle cell disease, first introduced the distinction between sickle cell disease and trait in 1933, although until 1949, the genetic characteristics had not been elucidated by James V. Neel and E.A. Beet. 1949 was the year when Linus Pauling described the unusual chemical behaviour of haemoglobin S, and attributed this to an abnormality in the molecule itself. The molecular change in HbS was described in 1956 by Vernon Ingram. The late 1940s and early 1950s saw further understanding in the link between malaria and sickle cell disease. In 1954, the introduction of haemoglobin electrophoresis allowed the discovery of particular subtypes, such as HbSC disease.
Large-scale natural history studies and further intervention studies were introduced in the 1970s and 1980s, leading to widespread use of prophylaxis against pneumococcal infections amongst other interventions. Bill Cosby's Emmy-winning 1972 TV movie, To All My Friends on Shore, depicted the story of the parents of a child with sickle cell disease. The 1990s had the development of hydroxycarbamide, and reports of cure through bone marrow transplantation appeared in 2007.
Some old texts refer to it as drepanocytosis.
Society and culture
United States
Effective 15 September 2017, the U.S. Social Security Administration issued a Policy Interpretation Ruling providing background information on sickle cell disease and a description of how Social Security evaluates the disease during its adjudication process for disability claims.
In the U.S., there are stigmas surrounding SCD that discourage people with SCD from receiving necessary care. These stigmas mainly affect people of African American and Latin American ancestries, according to the National Heart, Lung, and Blood Institute. People with SCD experience the impact of stigmas of the disease on multiple aspects of life including social and psychological well-being. Studies have shown that those with SCD frequently feel as though they must keep their diagnosis a secret to avoid discrimination in the workplace and also among peers in relationships. In the 1960s, the US government supported initiatives for workplace screening for genetic diseases in an attempt to be protective towards people with SCD. By having this screening, it was intended that employees would not be placed in environments that could potentially be harmful and trigger SCD.
Uganda
Uganda has the 5th highest sickle cell disease (SCD) burden in the world. In Uganda, social stigma exists for those with sickle cell disease because of the lack of general knowledge of the disease. The general gap in knowledge surrounding sickle cell disease is noted among adolescents and young adults due to the culturally sanctioned secrecy about the disease. While most people have heard generally about the disease, a large portion of the population is relatively misinformed about how SCD is diagnosed or inherited. Those who are informed about the disease learned about it from family or friends and not from health professionals. Failure to provide the public with information about sickle cell disease results in a population with a poor understanding of the causes of the disease, symptoms, and prevention techniques. The differences, physically and socially, that arise in those with sickle cell disease, such as jaundice, stunted physical growth, and delayed sexual maturity, can also lead them to become targets of bullying, rejection, and stigma.
Rate of sickle cell disease in Uganda
The data compiled on sickle cell disease in Uganda has not been updated since the early 1970s. The deficiency of data is due to a lack of government research funds, even though Ugandans die daily from SCD. Data shows that the trait frequency of sickle cell disease is 20% of the population in Uganda. This means that 66 million people are at risk of having a child who has sickle cell disease. It is also estimated that about 25,000 Ugandans are born each year with SCD and 80% of those people don't live past five years old. SCD also contributes 25% to the child mortality rate in Uganda. The Bamba people of Uganda, located in the southwest of the country, carry 45% of the gene which is the highest trait frequency recorded in the world. The Sickle Cell Clinic in Mulago is only one sickle cell disease clinic in the country and on average sees 200 patients a day.
Misconceptions about sickle cell disease
The stigma around the disease is particularly bad in regions of the country that are not as affected. For example, Eastern Ugandans tend to be more knowledgeable of the disease than Western Ugandans, who are more likely to believe that sickle cell disease resulted as a punishment from God or witchcraft. Other misconceptions about SCD include the belief that it is caused by environmental factors but, in reality, SCD is a genetic disease. There have been efforts throughout Uganda to address the social misconceptions about the disease. In 2013, the Uganda Sickle Cell Rescue Foundation was established to spread awareness of sickle cell disease and combat the social stigma attached to the disease. In addition to this organization's efforts, there is a need for the inclusion of sickle cell disease education in preexisting community health education programs in order to reduce the stigmatization of sickle cell disease in Uganda.
Social isolation of people with sickle cell disease
The deeply rooted stigma of SCD from society causes families to often hide their family members' sick status for fear of being labeled, cursed, or left out of social events. Sometimes in Uganda, when it is confirmed that a family member has sickle cell disease, intimate relationships with all members of the family are avoided. The stigmatization and social isolation people with sickle cell disease tend to experience is often the consequence of popular misconceptions that people with SCD should not socialize with those free from the disease. This mentality robs people with SCD of the right to freely participate in community activities like everyone else SCD-related stigma and social isolation in schools, especially, can make a life for young people living with sickle cell disease extremely difficult. For school-aged children living with SCD, the stigma they face can lead to peer rejection. Peer rejection involves the exclusion from social groups or gatherings. It often leads the excluded individual to experience emotional distress and may result in their academic underperformance, avoidance of school, and occupational failure later in life. This social isolation is also likely to negatively impact people with SCD's self-esteem and overall quality of life.
Mothers of children with sickle cell disease tend to receive disproportionate amounts of stigma from their peers and family members. These women will often be blamed for their child's diagnosis of SCD, especially if SCD is not present in earlier generations, due to the suspicion that the child's poor health may have been caused by the mother's failure to implement preventative health measures or promote a healthy environment for her child to thrive. The reliance on theories related to environmental factors to place blame on the mother reflects many Ugandan's poor knowledge of how the disease is acquired as it is determined by genetics, not environment. Mothers of children with sickle cell disease are also often left with very limited resources to safeguard their futures against the stigma of having SCD. This lack of access to resources results from their subordinating roles within familial structures as well as the class disparities that hinder many mothers' ability to satisfy additional childcare costs and responsibilities.
Women living with SCD who become pregnant often face extreme discrimination and discouragement in Uganda. These women are frequently branded by their peers as irresponsible for having a baby while living with sickle cell disease or even engaging in sex while living with SCD. The criticism and judgement these women receive, not only from healthcare professionals but also from their families, often leaves them feeling alone, depressed, anxious, ashamed, and with very little social support. Most pregnant women with SCD also go on to be single mothers as it is common for them to be left by their male partners who claim they were unaware of their partner's SCD status. Not only does the abandonment experienced by these women cause emotional distress for them, but this low level of parental support can be linked to depressive symptoms and overall lower quality of life for the child once they are born.
United Kingdom
In 2021 many patients were found to be afraid to visit hospitals so purchasing pain relief to treat themselves outside the NHS, such was the level of ignorance among staff. They were often waiting a long time for pain relief, and sometimes suspected of “drugs-seeking” behaviour. Delays to treatment, failure to inform the hospital haematology team and poor pain management had caused deaths. Specialist haematology staff prefer to work in bigger, teaching hospitals, leading to shortages of expertise elsewhere. In 2021 the NHS initiated its first new treatment in 20 years for Sickle Cell. This involved the use of Crizanlizumab, a drug given via transfusion drips, which reduces the number of visits to A and E by sufferers. The treatment can be accessed, via consultants, at any of ten new hubs set up around the country. In the same year, however, an All-Party Parliamentary Group produced a report on Sickle Cell and Thalassaemia entitled 'No-one is listening'. Partly in response to this, on 19 June 2022, World Sickle Cell Day, the NHS launched a campaign called " Can you tell it's sickle cell?". The campaign had twin aims. One was to increase awareness of the key signs and symptoms of the blood disorder so that people are as alert to signs of a sickle cell crisis as they are to an imminent heart attack or stroke. The second aim was to set up a new training programme to help paramedics, Accident and Emergency staff, carers and the general public to care effectively for sufferers in crisis.
Research
Umbilical cord blood transplant
While umbilical cord blood transplant can potentially cure the condition, a suitable donor is available in only 10% of people. About 7% of people also die as a result of the procedure and graft versus host disease may occur.
Gene therapy
Diseases such as sickle cell disease for which a person's normal phenotype or cell function may be restored in cells that have the disease by a normal copy of the gene that is mutated, may be a good candidate for gene therapy treatment. The risks and benefits related to gene therapy for sickle cell disease are not known.
In 2001, sickle cell disease reportedly had been successfully treated in mice using gene therapy. The researchers used a viral vector to make the mice—which have essentially the same defect that causes human sickle cell disease—express production of fetal haemoglobin (HbF), which an individual normally ceases to produce shortly after birth. In humans, using hydroxyurea to stimulate the production of HbF has been known to temporarily alleviate sickle cell disease symptoms. The researchers demonstrated that this gene therapy method is a more permanent way to increase therapeutic HbF production.
Phase 1 clinical trials of gene therapy for sickle cell disease in humans were started in 2014. The clinical trials will assess the safety of lentiviral vector-modified bone marrow for adults with severe sickle cell disease. As of 2020, however, no randomized controlled trials have been reported. A case report for the first person treated was published in March 2017, with a few more people being treated since then.
Gene editing platforms like CRISPR/Cas9 have been used to correct the disease-causing mutation in hematopoietic stem cells taken from a person with the condition. In July 2019 the gene-editing tool CRISPR was used to edit bone marrow cells from a person with SCD to boost fetal haemoglobin by inhibiting the BCL11A gene. A number of researchers have considered the ethical implications of SCD being one of the first potential applications of CRISPR technology, given the historical abuses and neglect of the African American community by the medical field.
In 2017 twelve clinical trials were focusing on gene therapy to treat sickle cell anemia. Of those 12 trials, four of them replaced the mutated HBB gene with a healthy one. Three trials used Mozobil, a medication used to treat types of cancer, to determine whether the increase of stem cells can be used for gene therapy. One trial focused on analyzing bone marrow samples from patients with sickle cell anemia. Another trial experimented with using umbilical cord blood from babies both with and without sickle cell anemia to develop gene therapy.
Hematopoietic stem cell transplantation
There is no strong medical evidence to determine the risks and potential benefits related to treating people with sickle cell disease with hematopoietic stem cell transplantations.
Further reading
|
Library resources about Sickle cell disease |
- Brown RT, ed. (2006). Comprehensive handbook of childhood cancer and sickle cell disease: a biopsychosocial approach. Oxford University Press. ISBN 978-0-19-516985-0.
- Hill SA (2003). Managing Sickle Cell Disease in Low-Income Families. Temple University Press. ISBN 978-1-59213-195-2.
- Serjeant GR, Serjeant BE (2001). Sickle Cell Disease. Oxford University Press. ISBN 978-0-19-263036-0.
- Tapper M (1999). In the blood: sickle cell anemia and the politics of race. University of Pennsylvania Press. ISBN 978-0-8122-3471-8.