
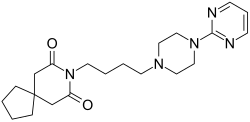
 | |
| Clinical data | |
|---|---|
| Pronunciation | /ˈbjuːspɪroʊn/ (BEW-spi-rohn) |
| Trade names | Buspar, Namanspin |
| Other names | MJ 9022-1 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a688005 |
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 3.9% |
| Protein binding | 86–95% |
| Metabolism | Liver (via CYP3A4) |
| Metabolites | 5-OH-Buspirone; 6-OH-Buspirone; 8-OH-Buspirone; 1-PP |
| Elimination half-life | 2.5 hours |
| Excretion |
Urine: 29–63% Feces: 18–38% |
| Identifiers | |
| |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.048.232 |
| Chemical and physical data | |
| Formula | C21H31N5O2 |
| Molar mass | 385.512 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| (verify) | |
Buspirone, sold under the brand name Buspar, among others, is a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder. Benefits support its short-term use. It is taken orally (by mouth), and takes two to six weeks to be fully effective.
Common side effects of buspirone include nausea, headaches, dizziness, and difficulty concentrating. Serious side effects may include movement disorders, serotonin syndrome, and seizures. Its use in pregnancy appears to be safe but has not been well studied, and use during breastfeeding has not been well studied. It is a serotonin 5-HT1A receptor agonist.
Buspirone was first made in 1968 and approved for medical use in the United States in 1986. It is available as a generic medication. In 2020, it was the 55th most-commonly prescribed medication in the United States, with more than 12 million prescriptions.
Medical uses
Anxiety
Buspirone is used for the short-term and long-term treatment of anxiety disorders or symptoms of anxiety. It is generally preferred over benzodiazepines because it does not activate the receptors that make drugs like alprazolam addictive.
Buspirone has no immediate anxiolytic effects, and hence has a delayed onset of action; its full clinical effectiveness may require 2–4 weeks to manifest itself. The drug has been shown to be similarly effective in the treatment of generalized anxiety disorder (GAD) to benzodiazepines including diazepam, alprazolam, lorazepam, and clorazepate. Buspirone is not known to be effective in the treatment of other anxiety disorders besides GAD, although there is some limited evidence that it may be useful in the treatment of social phobia as an adjunct to selective serotonin reuptake inhibitors (SSRIs).
Other uses
Sexual dysfunction
There is some evidence that buspirone on its own may be useful in the treatment of hypoactive sexual desire disorder (HSDD) in women. Buspirone may also be effective in treating antidepressant-induced sexual dysfunction.
Miscellaneous
Buspirone is not effective as a treatment for benzodiazepine withdrawal, barbiturate withdrawal, or alcohol withdrawal/delirium tremens.
SSRI and SNRI antidepressants such as paroxetine and venlafaxine may cause jaw pain/jaw spasm reversible syndrome (although it is not common), and buspirone appears to be successful in treating bruxism on SSRI/SNRI-induced jaw clenching.
Contraindications
Buspirone has these contraindications:
- Hypersensitivity to buspirone
- Metabolic acidosis, as in diabetes
- Should not be used with MAO inhibitors
- Severely compromised liver and/or kidney function
Side effects
Known side effects associated with buspirone include dizziness, headaches, nausea, tinnitus, and paresthesia. Buspirone is relatively well tolerated, and is not associated with sedation, cognitive and psychomotor impairment, muscle relaxation, physical dependence, or anticonvulsant effects. In addition, buspirone does not produce euphoria and is not a drug of abuse. Dyskinesia, akathisia, myoclonus, parkinsonism, and dystonia were reported associated with buspirone. It is unclear if there is a risk of tardive dyskinesia or other movement disorders with buspirone.
Overdose
Buspirone appears to be relatively benign in cases of single-drug overdose, although no definitive data on this subject appear to be available. In one clinical trial, buspirone was administered to healthy male volunteers at a dosage of 375 mg/day, and produced side effects including nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress. In early clinical trials, buspirone was given at dosages even as high as 2,400 mg/day, with akathisia, tremor, and muscle rigidity observed. Deliberate overdoses with 250 mg and up to 300 mg buspirone have resulted in drowsiness in about 50% of individuals. One death has been reported in a co-ingestion of 450 mg buspirone with alprazolam, diltiazem, alcohol, cocaine.
Interactions
Buspirone has been shown in vitro to be metabolized by the enzyme CYP3A4. This finding is consistent with the in vivo interactions observed between buspirone and these inhibitors or inducers of cytochrome P450 3A4 (CYP3A4), among others:
- Itraconazole: Increased plasma level of buspirone
- Rifampicin: Decreased plasma levels of buspirone
- Nefazodone: Increased plasma levels of buspirone
- Haloperidol: Increased plasma levels of buspirone
- Carbamazepine: Decreased plasma levels of buspirone
- Grapefruit: Significantly increases the plasma levels of buspirone. See grapefruit–drug interactions.
- Fluvoxamine: Moderately increase plasma levels of buspirone.
Elevated blood pressure has been reported when buspirone has been administered to patients taking monoamine oxidase inhibitors (MAOIs).
Pharmacology
Pharmacodynamics
| Site | Ki (nM) | Species | Ref |
|---|---|---|---|
| 5-HT1A | 3.98–214 21 (median) |
Human |
|
| 5-HT1B | >100,000 | Rat | |
| 5-HT1D | 22,000–42,700 | Human | |
| 5-HT2A | 138 759–1,300 |
Human Rat |
|
| 5-HT2B | 214 | Human | |
| 5-HT2C | 490 1,100–6,026 |
Human Rat/pig |
|
| 5-HT3 | >10,000 | Rat | |
| 5-HT4 | >10,000 | Rat | |
| 5-HT6 | 398 | Mouse | |
| 5-HT7 | 375–381 840 |
Rat Human |
|
| α1 | 1,000 | Rat | |
| α2 | 6,000 | Rat | |
| α2A | 7.3 (1-PP) | Human | |
| β | 8,800 | Rat | |
| D1 | 33,000 | Rat | |
| D2 | 484 240 |
Human Rat |
|
| D3 | 98 | Human | |
| D4 | 29 | Human | |
| mACh | 38,000 | Rat | |
| GABAA (BDZ) |
>100,000 | Rat | |
| Values are Ki (nM). The smaller the value, the more strongly the drug binds to the site. | |||
Buspirone acts as an agonist of the serotonin 5-HT1A receptor with high affinity. It is a partial agonist of both presynaptic 5-HT1A receptors, which are inhibitory autoreceptors, and postsynaptic 5-HT1A receptors. It is thought that the main effects of buspirone are mediated via its interaction with the presynaptic 5-HT1A receptor, thus reducing the firing of serotonin-producing neurons. Buspirone also has lower affinities for the serotonin 5-HT2A, 5-HT2B, 5-HT2C, 5-HT6, and 5-HT7 receptors.
In addition to binding to serotonin receptors, buspirone is an antagonist of the dopamine D2 receptor with weak affinity. It preferentially blocks inhibitory presynaptic D2 autoreceptors, and antagonizes postsynaptic D2 receptors only at higher doses. In accordance, buspirone has been found to increase dopaminergic neurotransmission in the nigrostriatal pathway at low doses, whereas at higher doses, postsynaptic D2 receptors are blocked and antidopaminergic effects such as hypoactivity and reduced stereotypy, though notably not catalepsy, are observed in animals. Buspirone has also been found to bind with much higher affinity to the dopamine D3 and D4 receptors, where it is similarly an antagonist.
A major metabolite of buspirone, 1-(2-pyrimidinyl)piperazine (1-PP), occurs at higher circulating levels than buspirone itself and is known to act as a potent α2-adrenergic receptor antagonist. This metabolite may be responsible for the increased noradrenergic and dopaminergic activity observed with buspirone in animals. In addition, 1-PP may play an important role in the antidepressant effects of buspirone. Buspirone also has very weak and probably clinically unimportant affinity for the α1-adrenergic receptor. However, buspirone has been reported to have shown "significant and selective intrinsic efficacy" at the α1-adrenergic receptor expressed in a "tissue- and species-dependent manner".
Unlike benzodiazepines, buspirone does not interact with the GABAA receptor complex.
Pharmacokinetics
Buspirone has a low oral bioavailability of 3.9% relative to intravenous injection due to extensive first-pass metabolism. The time to peak plasma levels following ingestion is 0.9 to 1.5 hours. It is reported to have an elimination half-life of 2.8 hours, although a review of 14 studies found that the mean terminal half-life ranged between 2 and 11 hours, and one study even reported a terminal half-life of 33 hours. Buspirone is metabolized primarily by CYP3A4, and prominent drug interactions with inhibitors and inducers of this enzyme have been observed. Major metabolites of buspirone include 5-hydroxybuspirone, 6-hydroxybuspirone, 8-hydroxybuspirone, and 1-PP. 6-Hydroxybuspirone has been identified as the predominant hepatic metabolite of buspirone, with plasma levels that are 40-fold greater than those of buspirone after oral administration of buspirone to humans. The metabolite is a high-affinity partial agonist of the 5-HT1A receptor (Ki = 25 nM) similarly to buspirone, and has demonstrated occupancy of the 5-HT1A receptor in vivo. As such, it is likely to play an important role in the therapeutic effects of buspirone. 1-PP has also been found to circulate at higher levels than those of buspirone itself and may similarly play a significant role in the clinical effects of buspirone.

Chemistry
Buspirone is a member of the azapirone chemical class, and consists of azaspirodecanedione and pyrimidinylpiperazine components linked together by a butyl chain.
Analogues
Structural analogues of buspirone include other azapirones like gepirone, ipsapirone, perospirone, and tandospirone.
A number of analogues are recorded.
Synthesis
A number of more modern methods of synthesis have also been reported (list not exhaustive):
Alkylation of 1-(2-pyrimidyl)piperazine [20980-22-7] (1) with 3-chloro-1-cyanopropane (4-chlorobutyronitrile) [628-20-6] (2) gives [33386-14-0] (3). the reduction of the nitrile group is performed either by catalytic hydrogenation or with LAH giving [33386-20-8] (4). The primary amine is then reacted with 3,3-tetramethyleneglutaric anhydride [5662-95-3] (5) in order to yield Buspirone (6).
History
Buspirone was first synthesized by a team at Mead Johnson in 1968 but was not patented until 1980. It was initially developed as an antipsychotic acting on the D2 receptor but was found to be ineffective in the treatment of psychosis; it was then used as an anxiolytic instead. In 1986, Bristol-Myers Squibb gained FDA approval for buspirone in the treatment of GAD. The patent expired in 2001, and buspirone is now available as a generic drug.
Society and culture

Generic names
Buspirone is the INN, BAN, DCF, and DCIT of buspirone, while buspirone hydrochloride is its USAN, BANM, and JAN.
Brand name
Buspirone was primarily sold under the brand name Buspar. Buspar is currently listed as discontinued by the US Food and Drug Administration. In 2010, in response to a citizen petition, the US FDA determined that Buspar was not withdrawn from sale for reasons of safety or effectiveness.
2019 shortage
Due to interrupted production at a Mylan Pharmaceuticals plant in Morgantown, West Virginia, the United States experienced a shortage of buspirone in 2019.
Research
Some tentative research supports other uses such as the treatment of depression and behavioral problems following brain damage.
External links
-
 Media related to Buspirone at Wikimedia Commons
Media related to Buspirone at Wikimedia Commons - "Buspirone". Drug Information Portal. U.S. National Library of Medicine.