
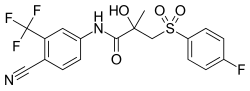
 | |
| Clinical data | |
|---|---|
| Routes of administration |
By mouth |
| Drug class | Nonsteroidal antiandrogen |
| Pharmacokinetic data | |
| Bioavailability | Well-absorbed; absolute bioavailability unknown |
| Protein binding |
Racemate: 96.1% (R)-Isomer: 99.6% (Mainly to albumin) |
| Metabolism |
Liver (extensively): • Hydroxylation (CYP3A4) • Glucuronidation (UGT1A9) |
| Metabolites | • Bicalutamide glucuronide • Hydroxybicalutamide • Hydroxybicalutamide gluc. (All inactive) |
| Elimination half-life | Single-dose: 6 days Continuous: 7–10 days |
| Excretion |
Feces: 43% Urine: 34% |
The pharmacology of bicalutamide, a nonsteroidal antiandrogen (NSAA), has been well-characterized. In terms of pharmacodynamics, bicalutamide acts as a selective antagonist of the androgen receptor (AR), the biological target of androgens like testosterone and dihydrotestosterone (DHT). It has no capacity to activate the AR. It does not decrease androgen levels and has no other important hormonal activity. The medication has progonadotropic effects due to its AR antagonist activity and can increase androgen, estrogen, and neurosteroid production and levels. This results in a variety of differences of bicalutamide monotherapy compared to surgical and medical castration, such as indirect estrogenic effects and associated benefits like preservation of sexual function and drawbacks like gynecomastia. Bicalutamide can paradoxically stimulate late-stage prostate cancer due to accumulated mutations in the cancer. When used as a monotherapy, bicalutamide can induce breast development in males due to its estrogenic effects. Unlike other kinds of antiandrogens, it may have less adverse effect on the testes and fertility.
In terms of pharmacokinetics, bicalutamide is well-absorbed when taken by mouth. However, absorption diminishes at higher dosages. It reaches maximal constant levels after 4 to 12 weeks of therapy. Bicalutamide shows extensive plasma protein binding, mainly to albumin. It crosses the blood–brain barrier and exerts effects in the central nervous system. Bicalutamide is metabolized in the liver by hydroxylation and glucuronidation. The metabolites of bicalutamide are not known to be active. The medication has a very long biological half-life of 6 days with a single dose and 7 to 10 days with repeated administration. Bicalutamide and its metabolites are eliminated in urine, feces, and bile, mainly in the form of conjugates. The pharmacokinetics of bicalutamide are not influenced by food, age, body weight, renal impairment, or mild-to-moderate hepatic impairment, but ethnicity may influence its pharmacokinetics in some cases.
Pharmacodynamics
| Antiandrogen | AR | PR-B | |||
|---|---|---|---|---|---|
| Ki (nM) | IC50 (nM) | Imax (%) | IC50 (nM) | Imax (%) | |
| Bicalutamide | 117 | 157 | 78 | 1,819 | 88 |
| Cyproterone acetate | 14 | 26 | 48 | >10,000 | 12 |
| Hydroxyflutamide | 27 | 15 | 83 | 2,013 | 90 |
| Mifepristone | 22 | 5 | 75 | 0.18 | 96 |
| Notes: IC50 values are for functional antagonism. Imax is maximal inhibition. Source: | |||||
| Compound | AR | PR | ER | GR | MR |
|---|---|---|---|---|---|
| Bicalutamide | 14–54 | 3,500–7,200 | >1,000,000 | 44,000–320,000 | ≥360,000 |
| Dihydrotestosterone | 0.5–3.1 | 280–440 | 38,000–340,000 | 2,700–20,000 | 2,100–2,300 |
| Notes: Values are Ki or IC50 (nM) for binding inhibition (affinity). Sources: | |||||
Antiandrogenic activity

| Compound | RBA |
|---|---|
| Metribolone | 100 |
| Dihydrotestosterone | 85 |
| Cyproterone acetate | 7.8 |
| Bicalutamide | 1.4 |
| Nilutamide | 0.9 |
| Hydroxyflutamide | 0.57 |
| Flutamide | <0.0057 |
|
Notes: | |
| Antiandrogen | Relative potency |
|---|---|
| Bicalutamide | 4.3 |
| Hydroxyflutamide | 3.5 |
| Flutamide | 3.3 |
| Cyproterone acetate | 1.0 |
| Zanoterone | 0.4 |
| Description: Relative potencies of orally administered antiandrogens in antagonizing 0.8 to 1.0 mg/kg s.c. testosterone propionate-induced ventral prostate weight increase in castrated immature male rats. Higher values mean greater potency. Sources: See template. | |

Bicalutamide acts as a highly selective competitive silent antagonist of the androgen receptor (AR) (IC50 = 159–243 nM), the major biological target of the androgen sex hormones testosterone and dihydrotestosterone (DHT). It has no capacity to activate the AR under normal physiological circumstances. In addition to competitive antagonism of the AR, bicalutamide has been found to accelerate the degradation of the AR, and this action may also be involved in its activity as an antiandrogen. The activity of bicalutamide lies in the (R)-isomer, which binds to the AR with an affinity that is about 30-fold higher than that of the (S)-isomer. Levels of the (R)-isomer also notably are 100-fold higher than those of the (S)-isomer at steady-state.
In relation to its selectivity for the AR, unlike steroidal antiandrogens (SAAs) such as cyproterone acetate (CPA) and megestrol acetate, bicalutamide does not interact importantly with other steroid hormone receptors (including the ERs, PRs, GR, or MR), and in accordance, has no clinically relevant additional, off-target hormonal activity (estrogenic or antiestrogenic, progestogenic or antiprogestogenic, glucocorticoid or antiglucocorticoid, or mineralocorticoid or antimineralocorticoid). However, it has been reported that bicalutamide has weak affinity for the progesterone receptor (PR) (~100- to 500-fold lower than for the AR), where it acts as an antagonist (with only ~12-fold lower functional inhibition relative to the AR in one study). Hence, bicalutamide may have some antiprogestogenic activity, although the clinical relevance of this is unknown. Bicalutamide does not inhibit 5α-reductase and is not known to inhibit other enzymes involved in androgen steroidogenesis (e.g., CYP17A1). Although bicalutamide does not bind to the ERs, it can increase estrogen levels secondarily to blockade of the AR when used as a monotherapy in males, and for this reason, the medication can indirectly activate the ERs to a degree and hence have some indirect estrogenic effects in men. Also in contrast to SAAs, bicalutamide neither inhibits nor suppresses androgen production in the body (i.e., it does not act as an antigonadotropin or steroidogenesis inhibitor), and instead exclusively mediates its antiandrogen effects by blocking androgen binding and subsequent receptor activation at the level of the AR.
In addition to the classical nuclear AR, bicalutamide has also been identified as a potent antagonist of ZIP9, a membrane androgen receptor (mAR) and zinc transporter protein, with an IC50 of 66.3 nM (relative to Kd = 17.9 nM for testosterone). This protein appears to be involved in prostate cancer and breast cancer. Bicalutamide failed to affect testosterone signaling mediated by GPRC6A, another mAR, on the other hand.
| Species | IC50 (nM) | RBA (ratio) | ||||
|---|---|---|---|---|---|---|
| Bicalutamide | 2-Hydroxyflutamide | Nilutamide | Bica / 2-OH-flu | Bica / nilu | Ref | |
| Rat | 190 | 700 | ND | 4.0 | ND | |
| Rat | ~400 | ~900 | ~900 | 2.3 | 2.3 | |
| Rat | ND | ND | ND | 3.3 | ND | |
| Rata | 3595 | 4565 | 18620 | 1.3 | 5.2 | |
| Human | ~300 | ~700 | ~500 | 2.5 | 1.6 | |
| Human | ~100 | ~300 | ND | ~3.0 | ND | |
| Humana | 2490 | 2345 | 5300 | 1.0 | 2.1 | |
| Footnotes: a = Controversial data. Sources: See template. | ||||||
Drug levels, androgen levels, and efficacy
The affinity of bicalutamide for the AR is approximately 30 to 100 times lower than that of DHT (IC50 ≈ 3.8 nM), the main endogenous ligand of the receptor in the prostate gland. However, sufficiently high relative concentrations of bicalutamide (1,000- to 10,000-fold excess) are able to completely prevent activation of the AR by androgens like DHT and testosterone and subsequent upregulation of the transcription of androgen-responsive genes and associated effects. At steady-state, relative to the normal adult male range for testosterone levels (300–1,000 ng/dL), circulating concentrations of bicalutamide at 50 mg/day are roughly 600 to 2,500 times higher and at 150 mg/day around 1,500 to 8,000 times higher than circulating testosterone levels, while bicalutamide concentrations, relative to the mean testosterone levels present in men who have been surgically castrated (15 ng/dL), are approximately 42,000 times higher than testosterone levels at 50 mg/day.

Whereas testosterone is the major circulating androgen, DHT is the major androgen in the prostate gland.DHT levels in circulation are relatively low and only approximately 10% of those of circulating testosterone levels. Conversely, local concentrations of DHT in the prostate gland are 8- to 10-fold higher than circulating levels of DHT. This is due to high expression of 5α-reductase in the prostate gland, which very efficiently catalyzes the formation of DHT from testosterone such that over 90% of intraprostatic testosterone is converted into DHT. Relative to testosterone, DHT is 2.5- to 10-fold as potent as an AR agonist in bioassays, and hence, is a much stronger androgen in comparison. As such, AR signaling is exceptionally high in the prostate gland, and the effectiveness of bicalutamide monotherapy in the treatment of prostate cancer, which is roughly equivalent to that of gonadotropin-releasing hormone analogues (GnRH analogues), demonstrates a capacity of bicalutamide to strongly antagonize the AR. On the other hand, GnRH analogue monotherapy achieves only a 50 to 60% reduction in levels of DHT in the prostate gland, and combined androgen blockade (CAB), the combination of surgical castration or a GnRH analogue and bicalutamide, is significantly more effective than either modality alone in the treatment of prostate cancer. Bicalutamide monotherapy has been found to decrease circulating levels of prostate-specific antigen (PSA), a marker of prostate cancer growth, by 57% at 10 mg/day, 73% at 30 mg/day, 90% at 50 mg/day, 97% at 100 mg/day, and 97% at 150 mg/day, while a 97% reduction in PSA is observed with 50 mg/day bicalutamide as a part of CAB. It has also been reported that bicalutamide monotherapy decreases median circulating levels of PSA at 3 months by 86.7% at 100 mg/day, 91.1% at 150 mg/day, and 93.8% at 200 mg/day (relative to 94–97% for castration). Above a bicalutamide monotherapy dosage of 200 mg/day, up to 600 mg/day, decreases in PSA levels reach a plateau. In a study of very-high-dose bicalutamide monotherapy, decreases in PSA levels after 12 weeks were approximately 93% with 300 mg/day, 96% with 450 mg/day, 96% with 600 mg/day, and 96% with castration.
Earlier studies with bicalutamide instead assessed changes in prostatic acid phosphatase (PAP) levels; proportions of patients with decreases of PAP of greater than or equal to 50% were 33% with 10 mg/day, 53% with 30 mg/day, and 83% with 50 mg/day bicalutamide.PSA is a more sensitive and specific prostate cancer tumor marker than PAP and subsequent studies employed PSA.
Despite the high medication levels that are achieved, due to their relatively low affinities for the AR, it has been suggested that 5 to 10% of DHT may remain unblocked in the prostate gland with CAB using standard doses of first-generation NSAAs. In accordance, second-generation NSAAs like enzalutamide and apalutamide, which have 5- to 10-fold higher affinity for the AR than bicalutamide, have been found to be more effective than bicalutamide in the treatment of prostate cancer. However, in the TERRAIN and STRIVE trials, which compared bicalutamide and enzalutamide as a component of CAB and found that enzalutamide extended life by 3 to 4 times as much time as bicalutamide, the dosage of enzalutamide used (160 mg) was over 3 times that of the dosage of bicalutamide used (50 mg). As a result, it has been suggested that the 50 mg/day dosage of bicalutamide used in this study and in CAB in general may be suboptimal. This is in accordance with clinical findings that PSA decreases with CAB using bicalutamide plateau at a dosage of bicalutamide of 150 to 200 mg/day.
In women, total testosterone levels are 20-fold and free testosterone levels 40-fold lower relative to men. In addition, whereas bicalutamide monotherapy can increase testosterone levels by up to 2-fold in men, the medication does not increase testosterone levels in women. For these reasons, much lower dosages of bicalutamide (e.g., 25 mg/day in the hirsutism studies) may be used in women with significant antiandrogenic effectiveness.
Influences on hormone levels

| Dosage | Before | Aftera | Difference | Change |
|---|---|---|---|---|
| 10 mg/day | 400 ng/dL | 490–520 ng/dL | +90–120 ng/dL | +21–29% |
| 30 mg/day | 320 ng/dL | 490–550 ng/dL | +170–230 ng/dL | +55–73% |
| 50 mg/day | 370 ng/dL | 550–610 ng/dL | +180–240 ng/dL | +46–62% |
| 100 mg/day | 320 ng/dL | 460–490 ng/dL | +140–170 ng/dL | +45–55% |
| 150 mg/day | 290 ng/dL | 460–490 ng/dL | +170–200 ng/dL | +60–70% |
| 200 mg/day | 320 ng/dL | 520–550 ng/dL | +200–230 ng/dL | +64–73% |
| Footnotes: a = After 29 to 85 days of treatment. Sources: | ||||
In men, blockade of the AR by bicalutamide in the pituitary gland and hypothalamus prevents the negative feedback of androgens on the hypothalamic–pituitary–gonadal (HPG) axis, resulting in an increase in luteinizing hormone (LH) secretion and levels.Follicle-stimulating hormone (FSH) levels, in contrast, remain essentially unchanged. The increase in LH levels leads to an elevation in androgen and estrogen levels. At a dosage of 150 mg/day, bicalutamide has been found to increase testosterone levels by about 1.5- to 2-fold (59–97% increase) and estradiol levels by about 1.5- to 2.5-fold (65–146% increase). Levels of DHT are also increased to a lesser extent (by 24–30%), and concentrations of sex hormone-binding globulin (SHBG) and prolactin increase as well (by 8–42% and 40–65%, respectively) secondary to the increase in estradiol levels. The estradiol concentrations produced in men by bicalutamide monotherapy are said to approximate the low-normal estradiol levels of a premenopausal woman, while testosterone levels generally remain in the high end of the normal male range and rarely exceed it. Dosages of bicalutamide of 10 mg, 30 mg, and 50 mg per day have been found to produce a "moderate" effect on sex hormone levels in men with prostate cancer (notably providing indication that the drug has clinically-relevant antiandrogen effects in males at a dosage as low as 10 mg/day). The elevated levels of gonadotropins and gonadal steroids associated with NSAA monotherapy is a unique endocrine state which can be described as "hypergonadotropic hypergonadism".
Bicalutamide increases androgen and estrogen levels only in men, and does not do so in women. This is because androgen levels are comparatively far lower in women and in turn exert little to no basal suppression of the HPG axis. Minimal or no changes of importance in levels of total testosterone, free testosterone, dihydrotestosterone, estradiol, androstenedione (A4), dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEA-S), 3α-androstanediol glucuronide (3α-ADG), progesterone, 17α-hydroxyprogesterone (17α-OHP), LH, FSH, prolactin, or SHBG have been observed in women with hirsutism with or without polycystic ovary syndrome that were treated with 25 or 50 mg/day bicalutamide for 6 to 12 months. However, in one study in women with polycystic ovary syndrome, 25 mg/day bicalutamide significantly decreased levels of total and free testosterone and significantly increased levels of SHBG. In addition to the minimal changes in hormone levels in women, although bicalutamide monotherapy increases gonadotropin and sex hormone levels in men, this will not occur if bicalutamide is combined with an antigonadotropin such as a GnRH analogue, estrogen, or progestogen, as these medications maintain negative feedback on the HPG axis.
The reason that testosterone levels are elevated but almost always remain in the normal male range with bicalutamide monotherapy is thought to be due to the concomitantly increased levels of estradiol, as estradiol is potently antigonadotropic and limits secretion of LH. In fact, estradiol is a much stronger inhibitor of gonadotropin secretion than is testosterone, and even though circulating concentrations of estradiol are far lower than those of testosterone in men, it is said that estradiol is nonetheless likely the major feedback regulator of gonadotropin secretion in this sex. In accordance, clomifene, a selective estrogen receptor modulator with antiestrogenic activity, has been found to increase testosterone levels to as much as 250% of initial values in men with hypogonadism, and a study of clomifene treatment in normal men observed increases in FSH and LH levels of 70–360% and 200–700%, respectively, with increases in testosterone levels that were similar to the increases seen with the gonadotropins. In addition to systemic or circulating estradiol, local aromatization of testosterone into estradiol in the hypothalamus and pituitary gland may contribute to suppression of gonadotropin secretion.
Bicalutamide more than blocks the effects of the increased testosterone levels that it induces in men, which is evidenced by its dose-dependent antiandrogenic effects (e.g., PSA decreases) and by the fact that monotherapy with the drug is about as effective as GnRH analogue therapy in the treatment of prostate cancer. However, in contrast, the effects of the elevated estrogen levels remain unopposed by bicalutamide, and this is importantly involved in the feminizing side effects (e.g., gynecomastia) of the drug in men.
Testosterone levels decline with age in men and younger men have higher testosterone levels on average than older men. Men with prostate cancer treated with bicalutamide are relatively elderly. The increases in testosterone levels with NSAAs like flutamide and bicalutamide may result in greater absolute levels of testosterone and estradiol in younger men than in older men. In one study that administered flutamide, free testosterone levels in young men increased from about 26 pg/mL at baseline to about 34 pg/mL with flutamide (+31%) and in elderly men from about 16 pg/mL at baseline to about 21 pg/mL (+31%) with flutamide. Hence, free testosterone levels with flutamide were approximately 1.6-fold higher in young men than in elderly men in this study. In the case of estradiol, total estradiol levels in young men increased from about 26 pg/mL at baseline to about 45 pg/mL with flutamide (+73%) and in elderly men changed from about 31 pg/mL to about 30 pg/mL (–3%). Other studies have similarly found relatively high absolute testosterone levels with NSAAs in young males. For instance, one study administering flutamide to late-pubertal males found that total testosterone levels increased from 729 ng/dL at baseline to 991 ng/dL with flutamide (+34%).
![Testosterone levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer.[110]](//upload.wikimedia.org/wikipedia/commons/thumb/b/b7/Testosterone_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png/394px-Testosterone_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png)
Testosterone levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer.
![Testosterone levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer.[83]](//upload.wikimedia.org/wikipedia/commons/thumb/f/fa/Testosterone_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png/394px-Testosterone_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png)
Testosterone levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer.
![Estradiol levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer.[110]](//upload.wikimedia.org/wikipedia/commons/thumb/f/f6/Estradiol_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png/391px-Estradiol_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png)
Estradiol levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer.
![Estradiol levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer.[83]](//upload.wikimedia.org/wikipedia/commons/thumb/5/5a/Estradiol_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png/396px-Estradiol_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png)
Estradiol levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer.
![Testosterone levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer.[110]](http://upload.wikimedia.org/wikipedia/commons/thumb/b/b7/Testosterone_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png/394px-Testosterone_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png)
![Testosterone levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer.[83]](http://upload.wikimedia.org/wikipedia/commons/thumb/f/fa/Testosterone_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png/394px-Testosterone_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png)
![Estradiol levels with 10, 30, and 50 mg/day bicalutamide monotherapy in men with prostate cancer.[110]](http://upload.wikimedia.org/wikipedia/commons/thumb/f/f6/Estradiol_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png/391px-Estradiol_levels_with_10%2C_30%2C_and_50_mg_per_day_bicalutamide_monotherapy_in_men.png)
![Estradiol levels with 10 to 200 mg/day bicalutamide monotherapy in men with prostate cancer.[83]](http://upload.wikimedia.org/wikipedia/commons/thumb/5/5a/Estradiol_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png/396px-Estradiol_levels_with_10_to_200_mg_per_day_bicalutamide_monotherapy_in_men.png)
Differences from castration
It has been proposed that the increase in estrogen levels caused by NSAAs like bicalutamide compensates for androgen blockade in the brain, which may explain differences in the side effect profiles of these drugs relative to GnRH analogues/castration, combined androgen blockade, and CPA (which, in contrast, decrease both androgen and estrogen levels). In the case of sexual interest and function, this notion is supported by a variety of findings including animal studies showing that estrogen deficiency results in diminished sexual behavior, treatment with tamoxifen resulting in significantly lowered libido in 30% of men receiving it for male breast cancer, and estrogen administration restoring libido and the frequency of sexual intercourse in men with congenital estrogen deficiency, among others.
Several metabolites of testosterone and DHT, including estradiol, 3α-androstanediol, and 3β-androstanediol, are estrogens (mainly potent ERβ agonists in the cases of the latter two), and 3α-androstanediol is additionally a potent GABAA receptor-potentiating neurosteroid. Due to the fact that bicalutamide does not lower testosterone levels, the levels of these metabolites would not be expected to be lowered either, unlike with therapies such as GnRH analogues. (Indeed, testosterone, DHT, and estradiol levels are actually raised by bicalutamide treatment, and for this reason, levels of 3α- and 3β-androstanediol might be elevated to some degree similarly.) These metabolites of testosterone have been found to have AR-independent positive effects on sexual motivation, and may be involved in the preservation of sexual interest and function by bicalutamide and other NSAAs. However, a study found that a combination of bicalutamide and dutasteride, a 5α-reductase inhibitor and inhibitor of neurosteroid biosynthesis, produced fewer sexual side effects than GnRH analogue therapy, specifically suggesting the role of estradiol in the preservation sexual interest and function with bicalutamide monotherapy rather than of DHT metabolites.
As an alternative possibility to testosterone metabolites preserving sexual desire and function with bicalutamide, it has been suggested that bicalutamide may not be able to block the actions of androgens in the brain to a degree sufficient to cause substantial sexual impairment.
It has been reported that bicalutamide, surprisingly, has no antianabolic effect on testosterone propionate-induced increases in levator ani muscle weight at doses that inhibit and even completely prevent testosterone propionate-induced growth of the prostate gland and seminal vesicles in rats. As such, it has been said that, based on preclinical research, bicalutamide does not have marked antianabolic effects. However, higher doses of bicalutamide are able to considerably inhibit the growth of the levator ani muscle in rats. In any case, analogously to animal findings, high-dose bicalutamide monotherapy appears to preserve lean muscle mass and muscle strength in men with prostate cancer relative to GnRH agonists. It is notable that in contrast to castration, bicalutamide preserves and increases estrogen levels, and estrogens are thought to have positive effects on skeletal muscle, including on muscle mass.
Paradoxical stimulation of late-stage prostate cancer
Though a pure, or silent antagonist of the AR under normal circumstances, bicalutamide, as well as other earlier antiandrogens like flutamide and nilutamide, have been found to possess weak partial agonist properties in the setting of AR overexpression and agonist activity in the case of certain mutations in the ligand-binding domain (LBD) of the AR. As both of these circumstances can eventually occur in prostate cancer, resistance to bicalutamide usually develops and the drug has the potential to paradoxically stimulate tumor growth when this happens. This is the mechanism of the phenomenon of antiandrogen withdrawal syndrome, where antiandrogen discontinuation paradoxically slows the rate of tumor growth. The newer drug enzalutamide has been shown not to have agonistic properties in the context of overexpression of the AR, though certain mutations in the AR can still convert it from an antagonist to agonist.
Induction of breast development
| Study | N | Dosage | Gynecomastia | Breast tenderness | Ref |
|---|---|---|---|---|---|
| Tyrrell et al. (1998)a | 386 | 10 mg/day | 9% | 11% | |
| 30 mg/day | 26% | 42% | |||
| 50 mg/day | 36% | 48% | |||
| 100 mg/day | 79% | 86% | |||
| 150 mg/day | 78% | 89% | |||
| 200 mg/day | 79% | 79% | |||
| Kennealey & Furr (1991)b | 210 | 10 mg/day | 29% | 38% | |
| 30 mg/day | 60% | 64% | |||
| 50 mg/day | 52% | 60% | |||
| Zanardi et al. (2006)c | 66 | 0 mg/week (controls) | 0% | 0% | |
| 50 mg/week (~7 mg/day) | 44% | 32% | |||
| 100 mg/week (~14 mg/day) | 50% | 64% | |||
| Footnotes: a = Testosterone levels increased to ~460–610 ng/dL and estradiol levels to ~32–51 pg/mL. b = Testosterone levels increased to ~505–715 ng/dL and estradiol levels to ~32–53 pg/mL. c = Testosterone levels increased to ~540–600 ng/dL and estradiol levels to ~29–34 pg/mL. | |||||
In transgender women, breast development is a desired effect of antiandrogen and/or estrogen treatment. Bicalutamide induces breast development in individuals assigned male at birth by two mechanisms: 1) blocking androgen signaling in breast tissue; and 2) increasing estrogen levels. Estrogen is responsible for the induction of breast development under normal circumstances, while androgens powerfully suppress estrogen-induced breast growth. It has been found that very low levels of estrogen can induce breast development in the presence of low or no androgen signaling. In accordance, not only does bicalutamide induce gynecomastia at a high rate when given as a monotherapy to men with prostate cancer (38–85%; 66% in one very large trial), similarly to high-dose estrogen therapy with diethylstilbestrol (41–77%), NSAAs have been found to result in a higher incidence of gynecomastia in combination with a GnRH analogue (13–25%) relative to GnRH analogue therapy or castration alone (1–16%) (in spite of the presence of only castrate levels of estrogen in both cases). The rate of gynecomastia with CAB is also higher than with CPA monotherapy (7%).
A study of men treated with NSAA (flutamide or bicalutamide) monotherapy for prostate cancer found that NSAAs induced full ductal development and moderate lobuloalveolar development of the breasts from a histological standpoint. The study also found that, in contrast, treatment of transgender women with estrogen and CPA (which is progestogenic in addition to antiandrogenic, unlike NSAAs) resulted in full lobuloalevolar development, as well as pregnancy-like breast hyperplasia in two of the subjects. In addition, it was observed that the lobuloalveolar maturation reversed upon discontinuation of CPA after sex reassignment surgery in these individuals. It was concluded that progestogen in addition to antiandrogen/estrogen treatment is required for the induction of full female histological breast development (i.e., that includes complete lobuloalveolar maturation), and that continued progestogen treatment is necessary to maintain such maturation. It should be noted however that although these findings may have important implications in the contexts of lactation and breastfeeding, epithelial tissue accounts for approximately only 10% of breast volume (with the bulk of the breasts (80–90%) being represented by stromal or adipose tissue), and it is uncertain to what extent, if any, that development of lobuloalveolar structures (a form of epithelial tissue) contributes to breast size and/or shape.
Effects on spermatogenesis and fertility
Spermatogenesis and male fertility are dependent on FSH, LH, and high levels of testosterone within the testicles.LH does not seem to be involved in spermatogenesis outside of its role in inducing production of testosterone by the Leydig cells in the seminiferous tubules (which make up approximately 80% of the bulk of the testes), whereas this is not the case for FSH, which is importantly involved. In accordance with the fact that the testes are the source of 95% of circulating testosterone in the body, local levels of testosterone inside of the testes are extremely high, ranging from 20- to 200-fold higher than circulating concentrations. Moreover, high levels of testosterone within the testes are required for spermatogenesis, although only a small fraction (5–10%) of normal levels appears to actually be necessary for spermatogenesis.
Unlike with antigonadotropic antiandrogens like CPA and GnRH analogues, it has been reported that bicalutamide monotherapy (at 50 mg/day) has very little or no effect on the ultrastructure of the testes and on spermatogenesis in men even after long-term therapy (>4 years). This may be explained by the extremely high local levels of testosterone in the testes, in that it is likely that systemic bicalutamide therapy is unable to achieve concentrations of the drug within the testes that are able to considerably block androgen signaling in this part of the body. This is particularly so considering that bicalutamide increases circulating testosterone levels, and by extension gonadal testosterone production, by up to two-fold in males, and that only a small fraction of normal intratesticular testosterone levels, and by extension androgen action, appears to be necessary to maintain spermatogenesis. Bicalutamide monotherapy at 50 mg/day causes no or clinically unimportant Leydig cell hyperplasia.
In contrast to bicalutamide and other pure antiandrogens or NSAAs, antigonadotropic antiandrogens suppress gonadotropin secretion, which in turn diminishes testosterone production by the testes as well as the maintenance of the testes by FSH, resulting in atrophy and loss of their function. As such, bicalutamide and other NSAAs may uniquely have the potential to preserve testicular function and spermatogenesis and thus male fertility relative to alternative therapies. In accordance with this notion, a study found that prolonged, high-dose bicalutamide treatment had minimal effects on fertility in male rats. However, another study found that low-dose bicalutamide administration resulted in testicular atrophy and reduced the germ cell count in the testes of male rats by almost 50%, though the rate of successful fertilization and pregnancy following mating was not assessed. Additional studies found that bicalutamide decreased testes weights, altered testes histology, and decreased sperm count in male rats. Yet another study found that bicalutamide has no effect on testes weights or spermatogenesis in male rats.
Treatment of men with exogenous testosterone or other AAS results in suppression of gonadotropin secretion and gonadal testosterone production due to their antigonadotropic effects or activation of the AR in the pituitary gland, resulting in inhibition or abolition of spermatogenesis and fertility:
Treatment of an infertile man with testosterone does [not] improve spermatogenesis, since exogenous administrated testosterone and its metabolite estrogen will suppress both GnRH production by the hypothalamus and luteinizing hormone production by the pituitary gland and subsequently suppress testicular testosterone production. Also, high levels of testosterone are needed inside the testis and this can never be accomplished by oral or parenteral administration of androgens. Suppression of testosterone production by the leydig cells will result in a deficient spermatogenesis, as can be seen in men taking anabolic–androgenic steroids.
In contrast, pure AR antagonists would, in theory, result in the opposite (although reduced semen volume and sexual dysfunction may occur):
It is theoretically a sound hypothesis that the spermatogenesis can be increased by indirectly stimulating FSH and LH secretions from the pituitary gland. However, for this to fructify, it requires the use of testosterone antagonist to nullify the negative feedback effect of circulating testosterone on the release of FSH and LH, thus augmenting the secretion of testosterone and spermatogenesis. Unfortunately, a testosterone antagonist will be unacceptable to males, as it may reduce secondary sexual functions including erection and ejaculation that is vital for the successful fertilization.
However, while bicalutamide does not appear to adversely influence testicular spermatogenesis, and healthy sperm can be produced within the testes during bicalutamide monotherapy, AR antagonists may be able to interfere with male fertility via interference with androgen signaling beyond the testes. The maturation as well as transport of sperm occurs not only in the testes but also outside of the testes in the epididymides and vas deferens, and these processes in these tissues are dependent on AR signaling similarly to testicular spermatogenesis. However, whereas androgen levels are extremely high in the testes, this is not true in the epididymides and vas deferens. As androgen levels are relatively low in these tissues, at least compared to the testes, bicalutamide may be able to block AR signaling in these parts of the body to an extent that is sufficient to interfere with male fertility. Indeed, the AAS mesterolone has been used to improve sperm quality and fertility in men because, apparently unlike other AAS, it shows minimal antigonadotropic effects at typical clinical dosages but activates the AR and thereby supports sperm maturation in the epididymides. However, this use of mesterolone is controversial and its efficacy for such purposes is not fully certain.
Although bicalutamide alone would appear to have minimal detrimental effect on testicular spermatogenesis and hence on certain aspects of male fertility, other hormonal agents that bicalutamide may be combined with, including GnRH analogues and particularly estrogens (as in transgender hormone therapy), can have a considerable detrimental effect on fertility. This is largely a consequence of their antigonadotropic activity. Antigonadotropic agents like high-dose CPA, high-dose androgens (e.g., testosterone esters), and GnRH antagonists (though notably not GnRH agonists in the case of fertility) produce hypogonadism and high rates of severe or complete infertility (e.g., severe oligospermia or complete azoospermia) in men. However, these effects are fully and often rapidly reversible with their discontinuation, even after prolonged treatment. In contrast, while estrogens at sufficiently high dosages similarly are able to produce hypogonadism and to abolish or severely impair spermatogenesis, this is not necessarily reversible in the case of estrogens and can be long-lasting after prolonged exposure. The difference is attributed to an apparently unique, direct cytotoxic and adverse effect of high concentrations of estrogens on the Leydig cells of the testes.
Other activities
Cytochrome P450 modulation
It has been reported that bicalutamide may have the potential to inhibit the enzymes CYP3A4 and, to a lesser extent, CYP2C9, CYP2C19, and CYP2D6, based on in vitro research. However, no relevant inhibition of CYP3A4 has been observed in vivo with bicalutamide at a dose of 150 mg (using midazolam as a specific marker of CYP3A4 activity). In animals, bicalutamide has been found to be an inducer of certain cytochrome P450 enzymes. However, dosages of 150 mg/day or less have shown no evidence of this in humans.
Bicalutamide has been identified as a strong CYP27A1 (cholesterol 27-hydroxylase) inhibitor in vitro. CYP27A1 converts cholesterol into 27-hydroxycholesterol, an oxysterol that has multiple biological functions including direct, tissue-specific activation of the ER (it has been characterized as a selective estrogen receptor modulator) and the liver X receptor. 27-Hydroxycholesterol has been found to increase ER-positive breast cancer cell growth via its estrogenic action, and hence, it has been proposed that bicalutamide and other CYP27A1 inhibitors may be effective as adjuvant therapies to aromatase inhibitors in the treatment of ER-positive breast cancer. In addition to CYP27A1, bicalutamide has been found to bind to and inhibit CYP46A1 (cholesterol 24-hydroxylase) in vitro, but this has yet to be assessed and confirmed in vivo.
P-Glycoprotein inhibition
Bicalutamide, as well as enzalutamide, have been found to act as inhibitors of P-glycoprotein efflux and ATPase activity. This action may reverse docetaxel resistance in prostate cancer cells by reducing transport of the drug out of these cells.
GABAA receptor positive modulation
All of the NSAAs approved for the treatment of prostate cancer have been found to possess an off-target action of acting as weak non-competitive inhibitors of human GABAA receptor currents in vitro to varying extents. The IC50 values are 44 μM for flutamide (as hydroxyflutamide), 21 μM for nilutamide, 5.2 μM for bicalutamide, and 3.6 μM for enzalutamide. In addition, flutamide, nilutamide, and enzalutamide have been found to cause convulsions and/or death in mice at sufficiently high doses. Bicalutamide was notably not found to do this, but this was likely simply due to the limited central nervous system penetration of bicalutamide in this species. In any case, enzalutamide is the only approved NSAA that has been found to be associated with a significantly increased incidence of seizures and other associated side effects clinically, so the relevance of the aforementioned findings with regard to bicalutamide and the other NSAAs is unclear.
Miscellaneous
Bicalutamide has been identified as a potent antagonist of the protease-activated receptor 2 (PAR-2) and as a ligand and inhibitor of α2-macroglobulin.
Pharmacokinetics
| 50 mg/day | 150 mg/day | |
|---|---|---|
| Cmax | 0.77 μg/mL (1.8 μmol/L) |
1.4 μg/mL (3.3 μmol/L) |
| tmax | 31 hours | 39 hours |
| Css | 8.85 μg/mL (20.6 μmol/L) |
21.6–28.5 μg/mL (50.2–66.3 μmol/L) |
| tss | 4–12 weeks | 4–12 weeks |
| Notes: All values are for (R)-bicalutamide. Sources: | ||
The pharmacokinetics of bicalutamide are unaffected by food, age, body weight, renal impairment, and mild-to-moderate hepatic impairment. However, it has been observed that steady-state concentrations of bicalutamide are higher in Japanese individuals than in Caucasians, indicating that ethnicity may be associated with differences in the pharmacokinetics of bicalutamide in some instances.
Absorption
Bicalutamide is extensively and well-absorbed following oral administration, and its absorption is not affected by food. The absolute bioavailability of bicalutamide in humans is unknown due to its very low water solubility and hence lack of an assessable intravenous formulation. However, the absolute bioavailability of bicalutamide has been found to be high in animals at low doses (109% in mice at 10 mg/kg; 72% in rats at 1 mg/kg; 100% in dogs at 0.1 mg/kg), but diminishes with increasing doses such that the bioavailability of bicalutamide is low at high doses (10% in rats at 250 mg/kg; 31% in dogs at 100 mg/kg). In accordance, absorption of (R)-bicalutamide in humans is slow and extensive but saturable, with steady-state levels increasing linearly at a dosage of up to 150 mg/day and non-linearly at higher dosages.
At higher dosages of 100 to 200 mg/day, absorption of bicalutamide is approximately linear, with a small but increasing departure from linearity above 150 mg/day. In terms of geometric mean steady-state concentrations of (R)-bicalutamide, the departures from linearity were 4%, 13%, 17%, and 32% with dosages of 100, 150, 200, and 300 mg/day, respectively. There is a plateau in steady-state levels of (R)-bicalutamide with bicalutamide dosages above 300 mg/day, and, accordingly, dosages of bicalutamide of 300 to 600 mg/day result in similar circulating concentrations of (R)-bicalutamide and similar degrees clinically of efficacy, tolerability, and toxicity. Relative to 150 mg/day bicalutamide, levels of (R)-bicalutamide are about 15% higher at a dosage of 200 mg/day and about 50% higher at a dosage of 300 mg/day. In contrast to (R)-bicalutamide, the inactive enantiomer (S)-bicalutamide is much more rapidly absorbed (as well as cleared from circulation).
Steady-state concentrations of the drug are reached after 4 to 12 weeks of administration independently of dosage, with an approximate 10- to 20-fold progressive accumulation of circulating levels of (R)-bicalutamide. The relatively long time to reach steady-state is a product of the long elimination half-life of bicalutamide. With single 50 mg and 150 mg doses of bicalutamide, mean peak concentrations (Cmax) of (R)-bicalutamide are 0.77 μg/mL (1.8 μmol/L) (at 31 hours) and 1.4 μg/mL (3.3 μmol/L) (at 39 hours), respectively. At steady-state, mean circulating concentrations (Css) of (R)-bicalutamide with 50 mg/day and 150 mg/day bicalutamide are 8.85 μg/mL (20.6 μmol/L) and 21.6 μg/mL (50.2 μmol/L), respectively. In another 150 mg/day bicalutamide study, mean circulating concentrations of (R)-bicalutamide were 19.4 μg/mL (45.1 μmol/L) and 28.5 μg/mL (66.3 μmol/L) on days 28 and 84 (weeks 4 and 12) of treatment, respectively.
There is wide interindividual variability, up to 15.7-fold, in steady-state (R)-bicalutamide levels with bicalutamide therapy. This is the case for all dosage levels of bicalutamide, and ranges in (R)-bicalutamide levels for different dosages show significant overlap.
Distribution
The apparent oral volume of distribution (VSS/F) at steady state of (R)-bicalutamide with oral administration of a single 5 to 80 mg dose of (R)-bicalutamide in a novel solid dispersion with a polymeric matrix of hydroxypropyl methylcellulose phthalate (HP55S) (also known as (R)-bicalutamide/HP55S) ranges from 22.53 ± 3.71 L to 25.38 ± 2.69 L. Bicalutamide is highly protein-bound (96.1% for racemic bicalutamide, 99.6% for (R)-bicalutamide), mainly to albumin. It has negligible affinity for SHBG and no affinity for corticosteroid-binding globulin.
The tissue distribution of bicalutamide is not well-characterized. However, it has been reported that distribution studies with bicalutamide have shown that preferential (i.e., tissue-selective) accumulation in anabolic (e.g., muscle) tissues does not occur. There are no available data on hepatic bicalutamide concentrations in humans, but a rat study found that oral bicalutamide treatment resulted in 4-fold higher concentrations of the drug in the liver relative to plasma (a common finding with orally administered drugs, due to transfer through the hepatic portal system prior to reaching circulation). In men receiving 150 mg/day bicalutamide, concentrations of (R)-bicalutamide in semen were 4.9 μg/mL (11 μmol/L), and the amount of the drug that could potentially be delivered to a female partner during sexual intercourse is regarded as low (estimated at 0.3 μg/kg) and below the amount that is required to induce changes in the offspring of laboratory animals.
Based on animal research, it was initially thought that bicalutamide was unable to cross the blood–brain barrier into the central nervous system and hence would be a peripherally-selective antiandrogen in humans. This conclusion was drawn from the finding that bicalutamide does not increase LH or testosterone levels in multiple tested animal species (including rats and dogs).AR antagonists like flutamide normally do this by blocking ARs in the pituitary gland and hypothalamus in the brain and thereby disinhibiting the HPG axis. In humans however, bicalutamide has been found to increase LH and testosterone levels, and to a comparable extent relative to flutamide and nilutamide. This occurs to a significant extent at even a very low dosage of 10 mg/day bicalutamide. As such, it appears that there are species differences in the central penetration of bicalutamide and that the medication does indeed cross the blood–brain barrier and affect central function in humans. This is supported by potential side effects of bicalutamide, in spite of increased testosterone levels, like hot flashes and decreased sexual interest in men. However, a clinical study comparing bicalutamide and flutamide in men found that bicalutamide had less influence on the HPG axis than flutamide, suggesting that bicalutamide might have a limited degree of peripheral selectivity, at least compared to other NSAAs, in humans.
Bicalutamide has been identified as a substrate of P-glycoprotein and of the breast cancer resistance protein (BCRP), though not of the multidrug resistance-associated protein 1 (MRP1). This may be involved in tumor resistance to bicalutamide. P-Glycoprotein is also known to play a major role in excluding drugs from the brain due to efflux back across the blood–brain barrier.
Metabolism
The metabolism of bicalutamide is hepatic and stereoselective. The inactive (S)-enantiomer is metabolized mainly by glucuronidation and is rapidly cleared from circulation, while the active (R)-isomer is slowly hydroxylated and then glucuronidated. In accordance, the active (R)-enantiomer has a far longer elimination half-life than the (S)-isomer, and circulating levels of (R)-bicalutamide are 10- to 20-fold and 100-fold higher than those of (S)-bicalutamide after a single dose and at steady-state, respectively. (R)-Bicalutamide is almost exclusively metabolized via hydroxylation into (R)-hydroxybicalutamide by the cytochrome P450 enzyme CYP3A4. Bicalutamide is also glucuronidated by UGT1A9, a UDP-glucuronyltransferase, into bicalutamide glucuronide, and (R)-hydroxybicalutamide glucuronide is formed from the metabolism of (R)-hydroxybicalutamide by UGT1A9. Similar to the inactive (S)-enantiomer of bicalutamide, (R)-hydroxybicalutamide is glucuronidated and rapidly cleared from circulation. None of the metabolites of bicalutamide are known to be active. Following administration of bicalutamide, only low concentrations of the metabolites are detectable in blood plasma, while unchanged bicalutamide predominates. (R)-Bicalutamide has a long elimination half-life of 5.8 days with a single dose, and an elimination half-life of 7 to 10 days with repeated administration, which allows for convenient once-daily dosing of bicalutamide.
|
Bicalutamide metabolism in humans
|
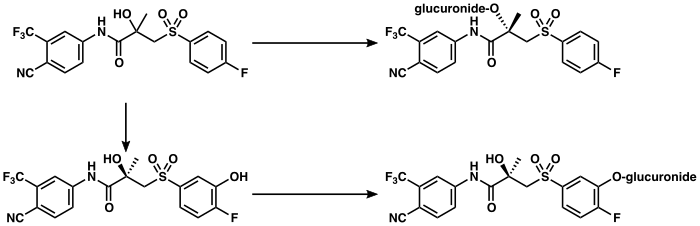
Elimination
Bicalutamide is eliminated in feces (43%) and urine (34%), whereas its metabolites are eliminated in approximately equal proportions in urine and bile. It is excreted to a substantial extent in its unmetabolized form, with both bicalutamide and its metabolites excreted mainly as glucuronide conjugates.
Graphs
![(R)-Bicalutamide levels after a single 5 to 80 mg dose of (R)-bicalutamide/HP55S in men.[191] The mean elimination half-life of (R)-bicalutamide in this study was 5.6 to 7.5 days.[191]](//upload.wikimedia.org/wikipedia/commons/thumb/4/43/%28R%29-Bicalutamide_levels_with_a_single_5_to_80_mg_oral_dose_of_bicalutamide_in_men.png/250px-%28R%29-Bicalutamide_levels_with_a_single_5_to_80_mg_oral_dose_of_bicalutamide_in_men.png)
(R)-Bicalutamide levels after a single 5 to 80 mg dose of (R)-bicalutamide/HP55S in men. The mean elimination half-life of (R)-bicalutamide in this study was 5.6 to 7.5 days.
![Bicalutamide levels after a single 50 mg dose of bicalutamide in men.[208] The mean elimination half-life of bicalutamide in this study was 4.2 days.[208]](//upload.wikimedia.org/wikipedia/commons/thumb/8/83/Bicalutamide_levels_after_a_single_50_mg_oral_dose_of_bicalutamide_in_men.png/250px-Bicalutamide_levels_after_a_single_50_mg_oral_dose_of_bicalutamide_in_men.png)
Bicalutamide levels after a single 50 mg dose of bicalutamide in men. The mean elimination half-life of bicalutamide in this study was 4.2 days.
![Bicalutamide levels after a single 10, 30, or 50 mg dose of bicalutamide in men.[209] The mean elimination half-life of bicalutamide in this study was 5.5 to 6.3 days.[209]](//upload.wikimedia.org/wikipedia/commons/thumb/c/cd/Bicalutamide_levels_after_a_single_10%2C_30%2C_or_50_mg_oral_dose_of_bicalutamide_in_men.png/250px-Bicalutamide_levels_after_a_single_10%2C_30%2C_or_50_mg_oral_dose_of_bicalutamide_in_men.png)
Bicalutamide levels after a single 10, 30, or 50 mg dose of bicalutamide in men. The mean elimination half-life of bicalutamide in this study was 5.5 to 6.3 days.
![Mean plasma (R)-bicalutamide concentrations with 10 to 600 mg/day bicalutamide in men over the course of 12 weeks.[83][65][210]](//upload.wikimedia.org/wikipedia/commons/thumb/5/5c/Mean_plasma_bicalutamide_levels_with_10_to_600_mg_per_day_bicalutamide_in_men.png/250px-Mean_plasma_bicalutamide_levels_with_10_to_600_mg_per_day_bicalutamide_in_men.png)
Mean plasma (R)-bicalutamide concentrations with 10 to 600 mg/day bicalutamide in men over the course of 12 weeks.
![Steady-state plasma levels of (R)-bicalutamide as a function of bicalutamide dosage (10 to 600 mg/day) in men.[83][65][210] Note the divergence from linearity at dosages above 200 mg/day, which demonstrates the saturation of absorption with higher dosages of bicalutamide.[210]](//upload.wikimedia.org/wikipedia/commons/thumb/0/06/Bicalutamide_levels_as_a_function_of_dosage_in_men.png/250px-Bicalutamide_levels_as_a_function_of_dosage_in_men.png)
Steady-state plasma levels of (R)-bicalutamide as a function of bicalutamide dosage (10 to 600 mg/day) in men. Note the divergence from linearity at dosages above 200 mg/day, which demonstrates the saturation of absorption with higher dosages of bicalutamide.
![(R)-Bicalutamide levels after a single 5 to 80 mg dose of (R)-bicalutamide/HP55S in men.[191] The mean elimination half-life of (R)-bicalutamide in this study was 5.6 to 7.5 days.[191]](http://upload.wikimedia.org/wikipedia/commons/thumb/4/43/%28R%29-Bicalutamide_levels_with_a_single_5_to_80_mg_oral_dose_of_bicalutamide_in_men.png/250px-%28R%29-Bicalutamide_levels_with_a_single_5_to_80_mg_oral_dose_of_bicalutamide_in_men.png)
![Bicalutamide levels after a single 50 mg dose of bicalutamide in men.[208] The mean elimination half-life of bicalutamide in this study was 4.2 days.[208]](http://upload.wikimedia.org/wikipedia/commons/thumb/8/83/Bicalutamide_levels_after_a_single_50_mg_oral_dose_of_bicalutamide_in_men.png/250px-Bicalutamide_levels_after_a_single_50_mg_oral_dose_of_bicalutamide_in_men.png)
![Bicalutamide levels after a single 10, 30, or 50 mg dose of bicalutamide in men.[209] The mean elimination half-life of bicalutamide in this study was 5.5 to 6.3 days.[209]](http://upload.wikimedia.org/wikipedia/commons/thumb/c/cd/Bicalutamide_levels_after_a_single_10%2C_30%2C_or_50_mg_oral_dose_of_bicalutamide_in_men.png/250px-Bicalutamide_levels_after_a_single_10%2C_30%2C_or_50_mg_oral_dose_of_bicalutamide_in_men.png)
![Mean plasma (R)-bicalutamide concentrations with 10 to 600 mg/day bicalutamide in men over the course of 12 weeks.[83][65][210]](http://upload.wikimedia.org/wikipedia/commons/thumb/5/5c/Mean_plasma_bicalutamide_levels_with_10_to_600_mg_per_day_bicalutamide_in_men.png/250px-Mean_plasma_bicalutamide_levels_with_10_to_600_mg_per_day_bicalutamide_in_men.png)
![Steady-state plasma levels of (R)-bicalutamide as a function of bicalutamide dosage (10 to 600 mg/day) in men.[83][65][210] Note the divergence from linearity at dosages above 200 mg/day, which demonstrates the saturation of absorption with higher dosages of bicalutamide.[210]](http://upload.wikimedia.org/wikipedia/commons/thumb/0/06/Bicalutamide_levels_as_a_function_of_dosage_in_men.png/250px-Bicalutamide_levels_as_a_function_of_dosage_in_men.png)