
 | |
 | |
| Clinical data | |
|---|---|
| Pronunciation | Bicalutamide: • /ˌbaɪkəˈluːtəmaɪd/ • BY-kə-LOO-tə-myde |
| Trade names | Casodex, Calutex, others |
| Other names | ICI-176,334; ZD-176,334 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a697047 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Nonsteroidal antiandrogen |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Well-absorbed; absolute bioavailability unknown |
| Protein binding |
Racemate: 96.1% (R)-Isomer: 99.6% (Mainly to albumin) |
| Metabolism |
Liver (extensively): • Hydroxylation (CYP3A4) • Glucuronidation (UGT1A9) |
| Metabolites | • Bicalutamide glucuronide • Hydroxybicalutamide • Hydroxybicalutamide gluc. (All inactive) |
| Elimination half-life | Single-dose: 5.8 days Continuous: 7–10 days |
| Excretion |
Feces: 43% Urine: 34% |
| Identifiers | |
| |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.126.100 |
| Chemical and physical data | |
| Formula | C18H14F4N2O4S |
| Molar mass | 430.37 g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture (of (R)- and (S)-enantiomers) |
| Melting point | 191 to 193 °C (376 to 379 °F) (experimental) |
| Boiling point | 650 °C (1,202 °F) (predicted) |
| Solubility in water | 0.005 |
| |
| |
| (verify) | |
Bicalutamide, sold under the brand name Casodex among others, is an antiandrogen medication that is primarily used to treat prostate cancer. It is typically used together with a gonadotropin-releasing hormone (GnRH) analogue or surgical removal of the testicles to treat metastatic prostate cancer (mPC). To a lesser extent, it is used at high doses for locally advanced prostate cancer (LAPC) as a monotherapy without castration. Bicalutamide was also previously used as monotherapy to treat localized prostate cancer (LPC), but authorization for this use was withdrawn following unfavorable trial findings. Besides prostate cancer, bicalutamide is limitedly used in the treatment of excessive hair growth and scalp hair loss in women, as a puberty blocker and component of feminizing hormone therapy for transgender girls and women, to treat gonadotropin-independent early puberty in boys, and to prevent overly long-lasting erections in men. It is taken by mouth.
Common side effects of bicalutamide in men include breast growth, breast tenderness, and hot flashes. Other side effects in men include feminization and sexual dysfunction. Some side effects like breast changes and feminization are minimal when combined with castration. While the medication appears to produce few side effects in women, its use in women is not explicitly approved by the Food and Drug Administration (FDA) at this time. Use during pregnancy may harm the baby. In men with early prostate cancer, bicalutamide monotherapy has been found to increase the likelihood of death from causes other than prostate cancer. Bicalutamide produces abnormal liver changes necessitating discontinuation in around 1% of people. Rarely, it has been associated with cases of serious liver damage, serious lung toxicity, and sensitivity to light. Although the risk of adverse liver changes is small, monitoring of liver function is recommended during treatment.
Bicalutamide is a member of the nonsteroidal antiandrogen (NSAA) group of medications. It works by selectively blocking the androgen receptor (AR), the biological target of the androgen sex hormones testosterone and dihydrotestosterone (DHT). It does not lower androgen levels. The medication can have some estrogen-like effects in men when used as a monotherapy due to increased estradiol levels. Bicalutamide is well-absorbed, and its absorption is not affected by food. The elimination half-life of the medication is around one week. It shows peripheral selectivity in animals, but crosses the blood–brain barrier and affects both the body and brain in humans.
Bicalutamide was patented in 1982 and approved for medical use in 1995. It is on the World Health Organization's List of Essential Medicines. Bicalutamide is available as a generic medication. The drug is sold in more than 80 countries, including most developed countries. It was at one time the most widely used antiandrogen in the treatment of prostate cancer, with millions of men with the disease having been prescribed it. Although bicalutamide is also used for other indications besides prostate cancer, the vast majority of prescriptions appear to be for treatment of prostate cancer.
Medical uses
Bicalutamide is approved for and mainly used in the following indications:
- Metastatic prostate cancer (mPC) in men in combination with a gonadotropin-releasing hormone (GnRH) analogue or surgical castration at 50 mg/day
- Locally advanced prostate cancer (LAPC) in men as a monotherapy at 150 mg/day (not approved for this use in the United States)
In Japan, bicalutamide is uniquely used at a dosage of 80 mg/day both in combination with castration and as a monotherapy in the treatment of prostate cancer.
Bicalutamide is also employed for the following off-label (non-approved) indications:
- To reduce the effects of the testosterone flare at the initiation of GnRH agonist therapy in men
- Androgen-dependent skin and hair conditions such as acne, seborrhea, excessive hair growth, and scalp hair loss in women as well as high testosterone levels due to polycystic ovary syndrome (PCOS) in women, at 25 to 50 mg/day generally in combination with a birth control pill
- Feminizing hormone therapy for transgender women in combination with an estrogen usually at 50 mg/day
- Peripheral precocious puberty in boys at 12.5 to 100 mg/day in combination with an aromatase inhibitor like anastrozole, especially for familial male-limited precocious puberty (testotoxicosis)
- Overly long-lasting erections in men at 50 mg per week to 50 mg every other day
The medication has been suggested for but has uncertain effectiveness in the following indication:
- Hypersexuality and paraphilias, particularly in combination with chemical castration
For more information on these uses, see the medical uses of bicalutamide article.
Available forms
Bicalutamide is available for the treatment of prostate cancer in most developed countries, including over 80 countries worldwide. It is available in 50 mg, 80 mg (in Japan), and 150 mg tablets for oral administration. The drug is registered for use as a 150 mg/day monotherapy for the treatment of LAPC in at least 55 countries, with the U.S. being a notable exception where it is registered only for use at a dosage of 50 mg/day in combination with castration. No other formulations or routes of administration are available or used. All formulations of bicalutamide are specifically indicated for the treatment of prostate cancer alone or in combination with surgical or medication castration. Due to the low water solubility of bicalutamide, bicalutamide in oral bicalutamide tablets is micronized to ensure small and consistent particle sizes and optimize oral bioavailability.
A combined formulation of bicalutamide and the GnRH agonist goserelin in which goserelin is provided as a subcutaneous implant for injection and bicalutamide is included as 50 mg tablets for oral ingestion is marketed in Australia and New Zealand under the brand name ZolaCos CP (Zoladex–Cosudex Combination Pack).
Contraindications
Bicalutamide is pregnancy category X, or "contraindicated in pregnancy", in the U.S., and pregnancy category D, the second most restricted rating, in Australia. As such, it is contraindicated in women during pregnancy, and women who are sexually active and who can or may become pregnant are strongly recommended to take bicalutamide only in combination with adequate contraception. It is unknown whether bicalutamide is excreted in breast milk, but many drugs are excreted in breast milk, and for this reason, bicalutamide treatment is similarly not recommended while breastfeeding.
In individuals with severe, though not mild-to-moderate hepatic impairment, there is evidence that the elimination of bicalutamide is slowed, and hence, caution may be warranted in these patients as circulating levels of bicalutamide may be increased. In severe hepatic impairment, the elimination half-life of the active (R)-enantiomer of bicalutamide is increased by about 1.75-fold (76% increase; elimination half-life of 5.9 and 10.4 days for normal and impaired patients, respectively). The elimination half-life of bicalutamide is unchanged in renal impairment.
Side effects
| Frequency | Class of effect | Effect |
|---|---|---|
| Very common (≥10%) | Reproductive system and breast disorders | • Breast tenderness • Gynecomastia |
| Common (1–10%) | General and psychiatric disorders | • Asthenia • Decreased libido • Erectile dysfunction • Hot flashes |
|
Skin and subcutaneous tissue disorders |
• Decreased body hair | |
| Hepato-biliary disorders | • Elevated liver enzymes | |
| Uncommon (0.1–1%) | Immune system disorders and hypersensitivity reactions | • Angioedema • Hives |
| Rare (<0.1%) or unknown | Respiratory disorders | • Lung disease |
| Skin and subcutaneous tissue disorders | • Sensitivity to light | |
| Hepato-biliary disorders | • Liver toxicity | |
The side effect profile of bicalutamide is highly dependent on sex; that is, on whether the person is male or female. In men, due to androgen deprivation, a variety of side effects of varying severity may occur during bicalutamide treatment, with breast pain/tenderness and gynecomastia (breast development/enlargement) being the most common. Gynecomastia occurs in up to 80% of men treated with bicalutamide monotherapy, and is of mild-to-moderate severity in more than 90% of affected men. In addition to breast changes, physical feminization and demasculinization in general, including reduced body hair growth, decreased muscle mass and strength, feminine changes in fat mass and distribution, reduced penile length, and decreased semen/ejaculate volume, may occur in men. Other side effects that have been observed in men and that are similarly related to androgen deprivation include hot flashes, sexual dysfunction (e.g., loss of libido, erectile dysfunction), depression, fatigue, weakness, and anemia. However, most men have preserved sexual function with bicalutamide monotherapy. In females, due to the minimal biological importance of androgens in this sex, the side effects of pure antiandrogens or NSAAs are few, and bicalutamide has been found to be very well tolerated. However, bicalutamide has been found to increase levels of total and LDL cholesterol in women. The non-pharmacological side-effect profile of bicalutamide (i.e., side effects not related to its antiandrogenic activity) is said to be similar to that with placebo. In any case, general side effects of bicalutamide that might occur in either sex include diarrhea, constipation, abdominal pain, nausea, dry skin, itching, and rash. The drug is well-tolerated at higher dosages than 50 mg/day, up to 600 mg/day, with rare additional side effects.
Bicalutamide has been associated with abnormal liver function tests such as elevated liver enzymes. In the Early Prostate Cancer (EPC) clinical programme of bicalutamide for LPC and LAPC, the rate of abnormal liver function tests with bicalutamide monotherapy was 3.4% relative to 1.9% for placebo. However, higher rates, up to 11%, have been seen in other studies.Hepatic changes that have necessitated discontinuation of bicalutamide, such as marked increases in liver enzymes or hepatitis, have occurred in 0.3 to 1.5% of men in clinical trials, or approximately 1% overall. Elevated liver enzymes with bicalutamide usually occur within the first 3 to 6 months of treatment. Monitoring of liver function during treatment is recommended, particularly in the first few months. In men with early prostate cancer, bicalutamide monotherapy has been found to increase non-prostate cancer mortality. The reasons for the increase in mortality with bicalutamide in these men are unknown, but possible factors could include androgen deprivation or drug-related toxicity of bicalutamide.
There are 10 published case reports of liver toxicity associated with bicalutamide as of 2022. Death occurred in 2 of these cases. Hundreds of additional cases of liver complications in people taking bicalutamide exist in the FDA Adverse Event Reporting System (FAERS) database. In all of the published case reports of liver toxicity with bicalutamide, the onset of symptoms was within the first 6 months of treatment. Symptoms that may indicate liver dysfunction include nausea, vomiting, abdominal pain, fatigue, anorexia, "flu-like" symptoms, dark urine, and jaundice. There are also published case reports of interstitial pneumonitis and eosinophilic lung disease associated with bicalutamide. along with hundreds of additional instances in the FAERS database as well. Interstitial pneumonitis can potentially progress to pulmonary fibrosis and may be fatal. Symptoms that may indicate lung dysfunction include dyspnea (difficult breathing or shortness of breath), cough, and pharyngitis (inflammation of the pharynx, resulting in sore throat). The exact incidence of liver toxicity and interstitial pneumonitis with bicalutamide are unknown, but both are said to be very rare events. A few cases of photosensitivity have been reported with bicalutamide.Hypersensitivity reactions (drug allergy) like angioedema and hives have also uncommonly been reported in association with bicalutamide.
Because it is an antiandrogen, bicalutamide has a theoretical risk of birth defects like ambiguous genitalia in male fetuses. Due to its teratogenic capacity, contraception should be used in women taking bicalutamide who are fertile and sexually active.
Comparison
The side effect profile of bicalutamide in men and women differs from that of other antiandrogens and is considered favorable in comparison. Relative to GnRH analogues and the steroidal antiandrogen (SAA) cyproterone acetate (CPA), bicalutamide monotherapy has a much lower incidence and severity of hot flashes and sexual dysfunction. In addition, unlike GnRH analogues and CPA, bicalutamide monotherapy is not associated with decreased bone mineral density or osteoporosis. Conversely, bicalutamide monotherapy is associated with much higher rates of breast tenderness, gynecomastia, and feminization in men than GnRH analogues and CPA. However, gynecomastia with bicalutamide is rarely severe and discontinuation rates due to this side effect are fairly low. These differences in side effects between bicalutamide monotherapy, GnRH analogues, and CPA are attributed to the fact that whereas GnRH analogues and CPA suppress estrogen production, bicalutamide monotherapy does not lower estrogen levels and in fact actually increases them.
Bicalutamide does not share the risk of neuropsychiatric side effects like fatigue as well as cardiovascular side effects like coagulation changes, blood clots, fluid retention, ischemic cardiomyopathy, and adverse serum lipid changes that CPA has been associated with. It has a much lower risk of hepatotoxicity than flutamide and CPA and of interstitial pneumonitis than nilutamide. The drug also does not share the unique risks of diarrhea with flutamide and nausea, vomiting, visual disturbances, and alcohol intolerance with nilutamide. Unlike enzalutamide, bicalutamide is not associated with seizures or related central side effects like anxiety and insomnia. However, although the risk of adverse liver changes with bicalutamide is low, enzalutamide differs from bicalutamide in having no known risk of elevated liver enzymes or hepatotoxicity. In contrast to the SAA spironolactone, bicalutamide does not have antimineralocorticoid effects, and hence is not associated with hyperkalemia, urinary frequency, dehydration, hypotension, or other related side effects. In women, unlike CPA and spironolactone, bicalutamide does not produce menstrual irregularity or amenorrhea and does not interfere with ovulation or fertility.
Overdose
A single oral dose of bicalutamide in humans that results in symptoms of overdose or that is considered to be life-threatening has not been established. Dosages of up to 600 mg/day have been well tolerated in clinical trials, and it is notable that there is a saturation of absorption with bicalutamide such that circulating levels of its active (R)-enantiomer do not further increase above a dosage of 300 mg/day. Overdose is considered unlikely to be life-threatening with bicalutamide or other first-generation NSAAs (i.e., flutamide and nilutamide). A massive overdose of nilutamide (13 grams, or 43 times the normal maximum 300 mg/day clinical dosage) in a 79-year-old man was uneventful, producing no clinical signs, symptoms, or toxicity. There is no specific antidote for bicalutamide or NSAA overdose, and treatment should be based on symptoms, if any are present.
Interactions
Bicalutamide is almost exclusively metabolized by CYP3A4. As such, its levels in the body may be altered by inhibitors and inducers of CYP3A4. (For a list of CYP3A4 inhibitors and inducers, see here.) However, in spite of the fact bicalutamide is metabolized by CYP3A4, there is no evidence of clinically significant drug interactions when bicalutamide at a dosage of 150 mg/day or less is co-administered with drugs that inhibit or induce cytochrome P450 enzyme activity.
In-vitro studies suggest that bicalutamide may be able to inhibit CYP3A4 and, to a lesser extent, CYP2C9, CYP2C19, and CYP2D6. Conversely, animal studies suggest that bicalutamide may induce cytochrome P450 enzymes. In a clinical study, bicalutamide co-administered with the CYP3A4 substrate midazolam caused only a small and statistically non-significant increase in midazolam levels (+27%) presumably due to CYP3A4 inhibition. However, this was well below increases in midazolam exposure with potent CYP3A4 inhibitors like ketoconazole (+1500%), itraconazole (+1000%), and erythromycin (+350%), and is considered to not be clinically important. There is no indication of clinically significant enzyme inhibition or induction with bicalutamide at doses of 150 mg/day or below.
Because bicalutamide circulates at relatively high concentrations and is highly protein-bound, it has the potential to displace other highly protein-bound drugs like warfarin, phenytoin, theophylline, and aspirin from plasma binding proteins. This could, in turn, result in increased free concentrations of such drugs and increased effects and/or side effects, potentially necessitating dosage adjustments. Bicalutamide has specifically been found to displace coumarin anticoagulants like warfarin from their plasma binding proteins (namely albumin) in vitro, potentially resulting in an increased anticoagulant effect, and for this reason, close monitoring of prothrombin time and dosage adjustment as necessary is recommended when bicalutamide is used in combination with these drugs. However, in spite of this, no conclusive evidence of an interaction between bicalutamide and other drugs was found in clinical trials of nearly 3,000 patients.
Pharmacology
Pharmacodynamics
Antiandrogenic activity
Bicalutamide acts as a highly selective competitive silent antagonist of the AR (IC50 = 159–243 nM), the major biological target of the androgen sex hormones testosterone and DHT, and hence is an antiandrogen. The activity of bicalutamide lies in the (R)-isomer. Due to its selectivity for the AR, bicalutamide does not interact importantly with other steroid hormone receptors and hence has no clinically relevant off-target hormonal activity (e.g., progestogenic, estrogenic, glucocorticoid, antimineralocorticoid). However, it has been reported that bicalutamide has weak affinity for the progesterone receptor (PR), where it is an antagonist, and hence it could have some antiprogestogenic activity. Bicalutamide does not inhibit 5α-reductase nor is known to inhibit other enzymes involved in androgen steroidogenesis (e.g., CYP17A1). Although it does not bind to the estrogen receptors (ERs), bicalutamide can increase estrogen levels secondarily to AR blockade when used as a monotherapy in males, and hence can have some indirect estrogenic effects in males. Bicalutamide neither suppresses nor inhibits androgen production in the body (i.e., it does not act as an antigonadotropin or androgen steroidogenesis inhibitor or lower androgen levels) and hence exclusively mediates its antiandrogenic effects by antagonizing the AR. In addition to the classical nuclear AR, bicalutamide has been assessed at the membrane androgen receptors (mARs) and found to act as a potent antagonist of ZIP9 (IC50 = 66.3 nM), whereas it does not appear to interact with GPRC6A.
The affinity of bicalutamide for the AR is relatively low as it is approximately 30 to 100 times lower than that of DHT, which is 2.5- to 10-fold as potent as an AR agonist as testosterone in bioassays and is the main endogenous ligand of the receptor in the prostate gland. However, typical clinical dosages of bicalutamide result in circulating levels of the drug that are thousands of times higher than those of testosterone and DHT, allowing it to powerfully prevent them from binding to and activating the receptor. This is especially true in the case of surgical or medical castration, in which testosterone levels in the circulation are approximately 95% reduced and DHT levels in the prostate gland are about 50 to 60% reduced. In women, levels of testosterone are substantially lower (20- to 40-fold) than in men, so much smaller doses of bicalutamide (e.g., 25 mg/day in the hirsutism studies) are necessary.
Blockade of the AR by bicalutamide in the pituitary gland and hypothalamus results in prevention of the negative feedback of androgens on the hypothalamic–pituitary–gonadal axis (HPG axis) in males and consequent disinhibition of pituitary luteinizing hormone (LH) secretion. This, in turn, results in an increase in circulating LH levels and activation of the gonadal production of testosterone and by extension production of estradiol. Levels of testosterone have been found to increase 1.5- to 2-fold (59–97% increase) and levels of estradiol about 1.5- to 2.5-fold (65–146% increase) in men treated with 150 mg/day bicalutamide monotherapy. In addition to testosterone and estradiol, there are smaller increases in concentrations of DHT, sex hormone-binding globulin, and prolactin. Estradiol levels with bicalutamide monotherapy are similar to those in the low-normal premenopausal female range while testosterone levels generally remain in the high end of the normal male range. Testosterone concentrations do not typically exceed the normal male range due to negative feedback on the HPG axis by the increased concentrations of estradiol. Bicalutamide influences the HPG axis and increases hormone levels only in men and not also in women. This is due to the much lower levels of androgens in women and their lack of basal suppression of the HPG axis in this sex. As evidenced by its effectiveness in the treatment of prostate cancer and other androgen-dependent conditions, the antiandrogenic actions of bicalutamide considerably exceed any impact of the increased levels of testosterone it results in. However, the elevated levels of estradiol remain unopposed by bicalutamide and are responsible for the gynecomastia and feminizing side effects it causes in men. Although bicalutamide monotherapy increases gonadotropin and sex hormone levels in men, this will not occur if bicalutamide is combined with an antigonadotropin such as a GnRH analogue, estrogen, or progestogen, as these medications maintain negative feedback on the HPG axis.
NSAA monotherapy, including with bicalutamide, shows a number of tolerability differences from methods of androgen deprivation therapy that incorporate surgical or medical castration. For example, the rates of hot flashes, depression, fatigue, and sexual dysfunction are all much higher with GnRH analogues than with NSAA monotherapy. It is thought that this is because GnRH analogues suppress estrogen production in addition to androgen production, resulting in estrogen deficiency. In contrast, NSAA monotherapy does not decrease estrogen levels and in fact increases them, resulting in an excess of estrogens that compensates for androgen deficiency and allows for a preservation of mood, energy, and sexual function.Neurosteroids that are produced from testosterone like 3α-androstanediol and 3β-androstanediol, which are ERβ agonists and the former a potent GABAA receptor positive allosteric modulator, may also be involved. In the specific case of sexual dysfunction, an additional possibility for the difference is that without concomitant suppression of androgen production, blockade of the AR by the bicalutamide in the brain is incomplete and insufficient to markedly influence sexual function.
Under normal circumstances, bicalutamide has no capacity to activate the AR. However, in prostate cancer, mutations and overexpression of the AR can accumulate in prostate gland cells which can convert bicalutamide from an antagonist of the AR into an agonist. This can result in paradoxical stimulation of prostate cancer growth with bicalutamide and is responsible for the phenomenon of the antiandrogen withdrawal syndrome, where antiandrogen discontinuation paradoxically slows the rate of prostate cancer growth.
In transgender women, breast development is a desired effect of antiandrogen or estrogen treatment. Breast development and gynecomastia induced by bicalutamide is thought to be mediated by increased activation of the ER secondary to blockade of the AR (resulting in disinhibition of the ER in breast tissue) and increased levels of estradiol. In addition to fat deposition, connective tissue growth, and ductal development, bicalutamide has been found to produce moderate lobuloalveolar development of the breasts. However, full lobuloalveolar maturation necessary for lactation and breastfeeding will not occur without progestogen treatment.
Bicalutamide monotherapy seems to have minimal effect on testicular spermatogenesis, testicular ultrastructure, and certain aspects of male fertility. This seems to be because testosterone levels in the testes (where ~95% of testosterone in males is produced) are extremely high (up to 200-fold higher than circulating levels) and only a small fraction (less than 10%) of the normal levels of testosterone in the testes are actually necessary to maintain spermatogenesis. As a result, bicalutamide appears to not be able to compete with testosterone in this sole part of the body to an extent sufficient to considerably interfere with androgen signaling and function. However, while bicalutamide does not seem to be able to adversely influence testicular spermatogenesis, it may interfere with AR-dependent sperm maturation and transport outside of the testes in the epididymides and vas deferens where androgen levels are far lower, and hence may still be able to impair male fertility. In addition, the combination of bicalutamide with other medications, such as estrogens, progestogens, and GnRH analogues, can compromise spermatogenesis due to their own adverse effects on male fertility. These medications are able to strongly suppress gonadal androgen production, which can severely impair or abolish testicular spermatogenesis, and estrogens also appear to have direct and potentially long-lasting cytotoxic effects in the testes at sufficiently high concentrations.
Other activities
Bicalutamide has been found to act as an inhibitor or inducer of certain cytochrome P450 enzymes including CYP3A4, CYP2C9, CYP2C19, and CYP2D6 in preclinical research, but no evidence of this has been found in humans treated with up to 150 mg/day. It has also been identified in vitro as a strong inhibitor of CYP27A1 (cholesterol 27-hydroxylase) and as an inhibitor of CYP46A1 (cholesterol 24-hydroxylase), but this has yet to be assessed or confirmed in vivo or in humans and the clinical significance remains unknown. Bicalutamide has been found to be a P-glycoprotein (ABCB1) inhibitor. Like other first-generation NSAAs and enzalutamide, it has been found to act as a weak non-competitive inhibitor of GABAA receptor-mediated currents in vitro (IC50 = 5.2 μM). However, unlike enzalutamide, bicalutamide has not been found to be associated with seizures or other related adverse central effects, so the clinical relevance of this finding is uncertain.
Pharmacokinetics
Though its absolute bioavailability in humans is unknown, bicalutamide is known to be extensively and well-absorbed. Its absorption is not affected by food. The absorption of bicalutamide is linear at doses up to 150 mg/day and is saturable at doses above this, with no further increases in steady-state levels of bicalutamide occurring at doses above 300 mg/day. Whereas absorption of (R)-bicalutamide is slow, with levels peaking at 31 to 39 hours after a dose, (S)-bicalutamide is much more rapidly absorbed. Steady-state concentrations of the drug are reached after 4 to 12 weeks of treatment independently of dosage, with a 10- to 20-fold progressive accumulation in levels of (R)-bicalutamide. The long time to steady-state levels is the result of bicalutamide's very long elimination half-life. There is wide interindividual variability in (R)-bicalutamide levels (up to 16-fold) with bicalutamide regardless of dosage.
The tissue distribution of bicalutamide is not well-characterized. The amount of bicalutamide in semen that could potentially be transferred to a female partner during sexual intercourse is low and is not thought to be important. Based on animal studies with rats and dogs it was thought that bicalutamide could not cross the blood–brain barrier and hence could not enter the brain. As such, it was initially thought to be a peripherally selective antiandrogen. However, subsequent clinical studies found that this was not also the case for humans, indicating species differences; bicalutamide crosses into the human brain and, in accordance, produces effects and side effects consistent with central antiandrogenic action. In any case, there is indication that bicalutamide might have at least some peripheral selectivity in humans. Bicalutamide is highly plasma protein bound (96.1% for racemic bicalutamide, 99.6% for (R)-bicalutamide) and is bound mainly to albumin, with negligible binding to SHBG and corticosteroid-binding globulin.
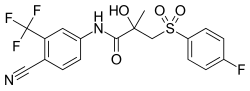
Bicalutamide is metabolized in the liver. (R)-Bicalutamide is metabolized slowly and almost exclusively via hydroxylation by CYP3A4 into (R)-hydroxybicalutamide. This metabolite is then glucuronidated by UGT1A9. In contrast to (R)-bicalutamide, (S)-bicalutamide is metabolized rapidly and mainly by glucuronidation (without hydroxylation). None of the metabolites of bicalutamide are known to be active and levels of the metabolites are low in plasma, where unchanged biclautamide predominates. Due to the stereoselective metabolism of bicalutamide, (R)-bicalutamide has a far longer terminal half-life than (S)-bicalutamide and its levels are about 10- to 20-fold higher in comparison following a single dose and 100-fold higher at steady-state. (R)-Bicalutamide has a relatively long elimination half-life of 5.8 days with a single dose and 7 to 10 days following repeated administration.
Bicalutamide is eliminated in similar proportions in feces (43%) and urine (34%), while its metabolites are eliminated roughly equally in urine and bile. The drug is excreted to a substantial extent in unmetabolized form, and both bicalutamide and its metabolites are eliminated mainly as glucuronide conjugates. The glucuronide conjugates of bicalutamide and its metabolites are eliminated from the circulation rapidly, unlike unconjugated bicalutamide.
The pharmacokinetics of bicalutamide are not affected by consumption of food, a person's age or body weight, renal impairment, or mild-to-moderate hepatic impairment. However, steady-state levels of bicalutamide are higher in Japanese individuals than in white people.
|
Bicalutamide metabolism in humans
|
Chemistry
Bicalutamide is a racemic mixture consisting of equal proportions of enantiomers (R)-bicalutamide (dextrorotatory) and (S)-bicalutamide (levorotatory). Its systematic name (IUPAC) is (RS)-N-[4-cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methylpropanamide. The compound has a chemical formula of C18H14F4N2O4S, a molecular weight of 430.373 g/mol, and is a fine white to off-white powder.
The acid dissociation constant (pKa') of bicalutamide is approximately 12. It is a highly lipophilic compound (log P = 2.92). At 37 °C (98.6 °F), or normal human body temperature, bicalutamide is practically insoluble in water (4.6 mg/L), acid (4.6 mg/L at pH 1), and alkali (3.7 mg/L at pH 8). In organic solvents, it is slightly soluble in chloroform and absolute ethanol, sparingly soluble in methanol, and freely soluble in acetone and tetrahydrofuran.
Bicalutamide is a synthetic and nonsteroidal compound which was derived from flutamide. It is a bicyclic compound (has two rings) and can be classified as and has variously been referred to as an anilide (N-phenylamide) or aniline, a diarylpropionamide, and a toluidide.
Analogues
First-generation NSAAs including bicalutamide, flutamide, and nilutamide are all synthetic, nonsteroidal anilide derivatives and structural analogues of each other. Bicalutamide is a diarylpropionamide while flutamide is a monoarylpropionamide and nilutamide is a hydantoin. Bicalutamide and flutamide, though not nilutamide, can also be classified as toluidides. All three of the compounds share a common 3-trifluoromethylaniline moiety. Bicalutamide is a modification of flutamide in which a 4-fluorophenylsulfonyl moiety has been added and the nitro group on the original phenyl ring has been replaced with a cyano group.Topilutamide, also known as fluridil, is another NSAA that is closely related structurally to the first-generation NSAAs, but, in contrast to them, is not used in the treatment of prostate cancer and is instead used exclusively as a topical antiandrogen in the treatment of pattern hair loss.
Bicalutamide




The second-generation NSAAs enzalutamide and apalutamide were derived from and are analogues of the first-generation NSAAs, while another second-generation NSAA, darolutamide, is said to be structurally distinct and chemically unrelated to the other NSAAs. Enzalutamide is a modification of bicalutamide in which the inter-ring linking chain has been altered and cyclized into a 5,5-dimethyl-4-oxo-2-thioxo imidazolidine moiety. In apalutamide, the 5,5-dimethyl groups of the imidazolidine ring of enzalutamide are cyclized to form an accessory cyclobutane ring and one of its phenyl rings is replaced with a pyridine ring.



The first nonsteroidal androgens, the arylpropionamides, were discovered via structural modification of bicalutamide. Unlike bicalutamide (which is purely antiandrogenic), these compounds show tissue-selective androgenic effects and were classified as selective androgen receptor modulators (SARMs). Lead SARMs of this series included acetothiolutamide, enobosarm (ostarine; S-22), and andarine (acetamidoxolutamide or androxolutamide; S-4). They are very close to bicalutamide structurally, with the key differences being that the linker sulfone of bicalutamide has been replaced with an ether or thioether group to confer agonism of the AR and the 4-fluoro atom of the pertinent phenyl ring has been substituted with an acetamido or cyano group to eliminate reactivity at the position.



A few radiolabeled derivatives of bicalutamide have been developed for potential use as radiotracers in medical imaging. They include [18F]bicalutamide, 4-[76Br]bromobicalutamide, and [76Br]bromo-thiobicalutamide. The latter two were found to have substantially increased affinity for the AR relative to that of bicautamide. However, none of these agents have been evaluated in humans.
5N-Bicalutamide, or 5-azabicalutamide, is a minor structural modification of bicalutamide which acts as a reversible covalent antagonist of the AR and has approximately 150-fold higher affinity for the AR and about 20-fold greater functional inhibition of the AR relative to bicalutamide. It is among the most potent AR antagonists to have been developed and is being researched for potential use in the treatment of antiandrogen-resistant prostate cancer.
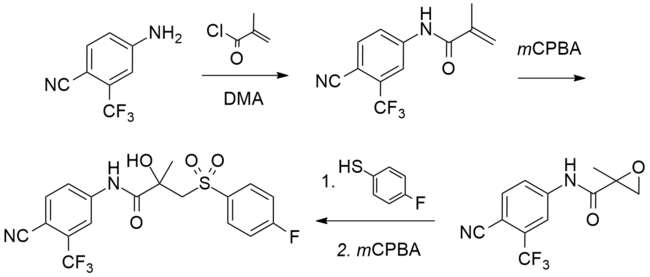
Synthesis
A number of chemical syntheses of bicalutamide have been published in the literature. The procedure of the first published synthesis of bicalutamide can be seen below.
|
Bicalutamide synthesis
|
History
Bicalutamide as well as all of the other currently marketed NSAAs were derived from structural modification of flutamide, which itself was originally synthesized as a bacteriostatic agent in 1967 at Schering Plough Corporation and was subsequently and serendipitously found to possess antiandrogenic activity. Bicalutamide was discovered by Tucker and colleagues at Imperial Chemical Industries (ICI) in the 1980s and was selected for development from a group of over 2,000 synthesized compounds. It was first patented in 1982 and was first reported in the scientific literature in June 1987.
Bicalutamide was first studied in a phase I clinical trial in 1987 and the results of the first phase II clinical trial in prostate cancer were published in 1990. The pharmaceutical division of ICI was split out into an independent company called Zeneca in 1993, and in April and May 1995, Zeneca (now AstraZeneca, after merging with Astra AB in 1999) began pre-approval marketing of bicalutamide for the treatment of prostate cancer in the U.S.. It was first launched in the U.K. in May 1995, and was subsequently approved by the U.S. FDA on 4 October 1995, for the treatment of prostate cancer at a dosage of 50 mg/day in combination with a GnRH analogue.
Following its introduction for use in combination with a GnRH analogue, bicalutamide was developed as a monotherapy at a dosage of 150 mg/day for the treatment of prostate cancer, and was approved for this indication in Europe, Canada, and a number of other countries in the late 1990s and early 2000s. This application of bicalutamide was also under review by the FDA in the U.S. in 2002, but ultimately was not approved in this country. In Japan, bicalutamide is licensed at a dosage of 80 mg/day alone or in combination with a GnRH analogue for prostate cancer. The unique 80 mg dosage of bicalutamide used in Japan was selected for development in this country on the basis of observed pharmacokinetic differences with bicalutamide in Japanese men.
Subsequent to negative findings of bicalutamide monotherapy for LPC in the EPC clinical programme, approval of bicalutamide for use specifically in the treatment of LPC was withdrawn in a number of countries including the U.K. (in October or November 2003) and several other European countries and Canada (in August 2003). In addition, the U.S. and Canada explicitly recommended against the use of 150 mg/day bicalutamide for this indication. The drug is effective for, remains approved for, and continues to be used in the treatment of LAPC and mPC, on the other hand.
The patent protection of bicalutamide expired in the U.S. in March 2009 and the drug has subsequently been available as a generic, at greatly reduced cost.
Bicalutamide was the fourth antiandrogen (and the third NSAA) to be introduced for the treatment of prostate cancer, following the SAA CPA in 1973 and the NSAAs flutamide in 1983 (1989 in the U.S.) and nilutamide in 1989 (1996 in the U.S.). It has been followed by abiraterone acetate in 2011, enzalutamide in 2012, apalutamide in 2018, and darolutamide in 2019, and may also be followed by in-development drugs such as proxalutamide and seviteronel.
Society and culture
Generic names
Bicalutamide is the generic name of the drug in English and French and its INN, USAN, USP,BAN, DCF, AAN, and JAN. It is also referred to as bicalutamidum in Latin, bicalutamida in Spanish and Portuguese, bicalutamid in German, and bikalutamid in Russian and other Slavic languages. The "bica-" prefix corresponds to the fact that bicalutamide is a bicyclic compound, while the "-lutamide" suffix is the standard suffix for NSAAs. Bicalutamide is also known by its former developmental code name ICI-176,334.
Brand names
Bicalutamide is marketed by AstraZeneca in oral tablet form under the brand names Casodex, Cosudex, Calutide, Calumid, and Kalumid in many countries. It is also marketed under the brand names Bicadex, Bical, Bicalox, Bicamide, Bicatlon, Bicusan, Binabic, Bypro, Calutol, and Ormandyl among others in various countries. The drug is sold under a large number of generic trade names such as Apo-Bicalutamide, Bicalutamide Accord, Bicalutamide Actavis, Bicalutamide Bluefish, Bicalutamide Kabi, Bicalutamide Sandoz, and Bicalutamide Teva as well. A combination formulation of bicalutamide and goserelin is marketed by AstraZeneca in Australia and New Zealand under the brand name ZolaCos-CP.
Cost and generics
Bicalutamide is off-patent and available as a generic. Unlike bicalutamide, the newer NSAA enzalutamide is still on-patent, and for this reason, is considerably more expensive in comparison.
The patent protection of all three of the first-generation NSAAs has expired and flutamide and bicalutamide are both available as low-cost generics. Nilutamide, on the other hand, has always been a poor third competitor to flutamide and bicalutamide and, in relation to this fact, has not been developed as a generic and is only available as brand name Nilandron, at least in the U.S.
Bicalutamide is considerably less costly than GnRH analogues, which, in spite of some having been off-patent many years, have been reported (in 2013) to typically cost US$10,000–$15,000 per year (or about US$1,000 per month) of treatment.
Sales and usage
Sales of bicalutamide (as Casodex) worldwide peaked at US$1.3 billion in 2007, and it has been described as a "billion-dollar-a-year" drug prior to losing its patent protection starting in 2007. In 2014, despite the introduction of abiraterone acetate in 2011 and enzalutamide in 2012, bicalutamide was still the most commonly prescribed drug in the treatment of metastatic castration-resistant prostate cancer (mCRPC). Moreover, in spite of being off-patent, bicalutamide was said to still generate a few hundred million dollars in sales per year for AstraZeneca. Total worldwide sales of brand name Casodex were approximately US$13.4 billion as of the end of 2018.
| Year | Sales | Year | Sales | Year | Sales | Year | Sales | Year | Sales | Year | Sales | Year | Sales | Year | Sales |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1995 | ~$15m | 1998 | $245m | 2001 | $569m | 2004 | $1012m | 2007* | $1335m | 2010 | $579m | 2013 | $376m | 2016 | $247m |
| 1996 | $109m | 1999 | $340m | 2002 | $644m | 2005 | $1123m | 2008 | $1258m | 2011 | $550m | 2014 | $320m | 2017 | $215m |
| 1997 | $200m | 2000 | $433m | 2003 | $854m | 2006 | $1206m | 2009 | $844m | 2012 | $454m | 2015 | $267m | 2018 | $201m |
| Notes: First generic availability (*) was in 2007. Total sales as of end 2018 were $13.4 billion. Sources: | |||||||||||||||
Between January 2007 and December 2009 (a period of three years), 1,232,143 prescriptions of bicalutamide were dispensed in the U.S., or about 400,000 prescriptions per year. During that time, bicalutamide accounted for about 87.2% of the NSAA market, while flutamide accounted for 10.5% of it and nilutamide for 2.3% of it. Approximately 96% of bicalutamide prescriptions were written for diagnosis codes that clearly indicated neoplasm. About 1,200, or 0.1% of bicalutamide prescriptions were dispensed to pediatric patients (age 0–16).
Regulation
Bicalutamide is a prescription drug. It is not specifically a controlled substance in any country and therefore is not an illegal drug. However, the manufacture, sale, distribution, and possession of prescription drugs are all still subject to legal regulation throughout the world.
Research
Bicalutamide has been studied in combination with the 5α-reductase inhibitors finasteride and dutasteride in prostate cancer. It has also been studied in combination with raloxifene, a selective estrogen receptor modulator (SERM), for the treatment of prostate cancer. Bicalutamide has been tested for the treatment of AR-positive ER/PR-negative locally advanced and metastatic breast cancer in women in a phase II study for this indication. Enzalutamide is also being investigated for this type of cancer. Bicalutamide has also been studied in a phase II clinical trial for ovarian cancer in women.
Bicalutamide has been studied in the treatment of benign prostatic hyperplasia (BPH) in a 24-week trial of 15 patients at a dosage of 50 mg/day. Prostate volume decreased by 26% in patients taking bicalutamide and urinary irritative symptom scores significantly decreased. Conversely, peak urine flow rates and urine pressure flow examinations were not significantly different between bicalutamide and placebo. The decrease in prostate volume achieved with bicalutamide was comparable to that observed with the 5α-reductase inhibitor finasteride, which is approved for the treatment of BPH. Breast tenderness (93%), gynecomastia (54%), and sexual dysfunction (60%) were all reported as side effects of bicalutamide at the dosage used in the study, although no treatment discontinuations due to adverse effects occurred and sexual functioning was maintained in 75% of patients.
A phase III clinical trial of bicalutamide in combination with an ethinylestradiol-containing combined oral contraceptive for the treatment of severe hirsutism in women with PCOS was completed in Italy in 2017 under supervision of the Italian Agency for Drugs (AIFA).
Antiandrogens have been suggested for treating COVID-19 in men and as of May 2020 high-dose bicalutamide is in a phase II clinical trial for this purpose.
Veterinary use
Bicalutamide may be used to treat hyperandrogenism and associated benign prostatic hyperplasia secondary to hyperadrenocorticism (caused by excessive adrenal androgens) in male ferrets. However, it has not been formally assessed in controlled studies for this purpose.
See also
Further reading
- "Bicalutamide". LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. NCBI Bookshelf. National Institute of Diabetes and Digestive and Kidney Diseases. 2017. PMID 31643303. NBK547970.
- Blackledge GR (1996). "Clinical progress with a new antiandrogen, Casodex (bicalutamide)". European Urology. 29 (Suppl 2): 96–104. doi:10.1159/000473847. PMID 8717470.
- Cockshott ID (2004). "Bicalutamide: clinical pharmacokinetics and metabolism". Clinical Pharmacokinetics. 43 (13): 855–78. doi:10.2165/00003088-200443130-00003. PMID 15509184.
- Fradet Y (February 2004). "Bicalutamide (Casodex) in the treatment of prostate cancer". Expert Review of Anticancer Therapy. 4 (1): 37–48. doi:10.1586/14737140.4.1.37. PMID 14748655. S2CID 34153031.
- Furr BJ (June 1995). "Casodex: preclinical studies and controversies". Annals of the New York Academy of Sciences. 761 (1): 79–96. Bibcode:1995NYASA.761...79F. doi:10.1111/j.1749-6632.1995.tb31371.x. PMID 7625752. S2CID 37242269.
- Furr BJ, Tucker H (January 1996). "The preclinical development of bicalutamide: pharmacodynamics and mechanism of action". Urology. 47 (1A Suppl): 13–25, discussion 29–32. doi:10.1016/S0090-4295(96)80003-3. PMID 8560673.
- Kolvenbag GJ, Blackledge GR (January 1996). "Worldwide activity and safety of bicalutamide: a summary review". Urology. 47 (1A Suppl): 70–9, discussion 80–4. doi:10.1016/s0090-4295(96)80012-4. PMID 8560681.
- Schellhammer PF, Davis JW (March 2004). "An evaluation of bicalutamide in the treatment of prostate cancer". Clinical Prostate Cancer. 2 (4): 213–9. doi:10.3816/CGC.2004.n.002. PMID 15072604.
- Tucker H, Crook JW, Chesterson GJ (1988). "Nonsteroidal antiandrogens. Synthesis and structure-activity relationships of 3-substituted derivatives of 2-hydroxypropionanilides". J. Med. Chem. 31 (5): 954–9. doi:10.1021/jm00400a011. PMID 3361581.
- Wellington K, Keam SJ (2006). "Bicalutamide 150mg: a review of its use in the treatment of locally advanced prostate cancer". Drugs. 66 (6): 837–50. doi:10.2165/00003495-200666060-00007. PMID 16706554. S2CID 46966712.