
| Selective estrogen receptor modulator | |
|---|---|
| Drug class | |
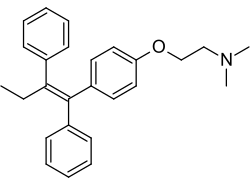
Tamoxifen, a nonsteroidal triphenylethylene antiestrogen and a widely used drug in the treatment of breast cancer.
| |
| Class identifiers | |
| Synonyms | SERM; Estrogen receptor agonist/antagonist; ERAA |
| Use | Breast cancer, infertility, osteoporosis, vaginal atrophy, dyspareunia, contraception, male hypogonadism, gynecomastia, breast pain, others |
| ATC code | G03XC |
| Biological target | Estrogen receptor |
| In Wikidata | |
Selective estrogen receptor modulators (SERMs), also known as estrogen receptor agonist/antagonists (ERAAs), are a class of drugs that act on the estrogen receptor (ER). A characteristic that distinguishes these substances from pure ER agonists and antagonists (that is, full agonists and silent antagonists) is that their action is different in various tissues, thereby granting the possibility to selectively inhibit or stimulate estrogen-like action in various tissues.
Medical uses
SERMs are used for various estrogen-related diseases, including treatment of ovulatory dysfunction in the management of infertility, treatment and prevention of postmenopausal osteoporosis, treatment and reduction in risk of breast cancer and treatment of dyspareunia due to menopause. SERM is also used in combination with conjugated estrogens indicated for the treatment of estrogen deficiency symptoms, and vasomotor symptoms associated with menopause. SERMs are used dependent on their pattern of action in various tissues:
Tamoxifen is a first-line hormonal treatment of ER-positive metastatic breast cancer. It is used for breast cancer risk reduction in women at high risk, and as adjuvant treatment of axillary node-negative and node-positive, ductal carcinoma in situ. Tamoxifen treatment is also useful in the treatment of bone density and blood lipids in postmenopausal women. Adverse effects include hot flushes and more serious is two to three times higher relative risk of developing endometrial cancer compared to women of an age-matched population.
Toremifene, a chlorinated tamoxifen derivative, causes fewer DNA adducts in liver than seen with tamoxifen in preclinical studies and was developed to avoid hepatic carcinomas. It is used as endocrine therapy in women with ER/PR-positive stage 4 or recurrent metastatic breast cancer and has demonstrated similar efficacy compared to tamoxifen as adjuvant treatment of breast cancer and in the treatment of metastatic breast cancer.
Raloxifene is used for prevention and treatment of postmenopausal osteoporosis and breast cancer prevention in high-risk postmenopausal women with osteoporosis. Preclinical and clinical reports suggest that it is considerably less potent than estrogen for the treatment of osteoporosis. It is associated with an acceptable endometrial profile and has not demonstrated tamoxifen-like effects in the uterus but has been associated with adverse effects such as venous thromboembolism and vasomotor symptoms, including hot flushes.
Ospemifene is an analogous metabolite of toremifene. Unlike tamoxifen, toremifene is not a rat hepatocarcinogen and therefore ospemifene would also be a safer SERM than tamoxifen. It is used for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy associated with menopause. Clinical data on breast cancer are not available, but both in vitro and in vivo data suggest that ospemifene may have chemopreventive activity in breast tissue.
Bazedoxifene is used as treatment for osteoporosis in postmenopausal women at increased risk of fracture. It has been shown to be relatively safe and well tolerated. It shows no breast or endometrial stimulation and in the first two years, the small increase is better in venous thromboembolism, and similar in the long term to other SERMs. The advantage of bazedoxifene over raloxifene is that it increases endothelial nitric oxide synthase activity and does not antagonize the effect of 17β-estradiol on vasomotor symptoms.
The first tissue selective estrogen complex (TSEC) combines conjugated estrogens and the SERM bazedoxifene to blend their activities. The combination therapy is used in the treatment of moderate to severe vasomotor symptoms associated with menopause, prevention of postmenopausal osteoporosis as well as treatment of estrogen deficiency symptoms in non-hysterectomized postmenopausal women. The combination allows for the benefits of estrogen with regard to relief of vasomotor symptoms without estrogenic stimulation of the endometrium.
Available forms
| Name | Brand name | Approved uses | Launch | Notes |
|---|---|---|---|---|
| Anordrin | Zi Yun | Emergency contraception | 1970s | Only in China, combined with mifepristone |
| Bazedoxifene | Duavee | Osteoporosis prevention | 2013 | Combined with conjugated estrogens |
| Broparestrol | Acnestrol | Dermatology; Breast cancer treatment | 1970s | Discontinued |
| Clomifene | Clomid | Female infertility | 1967 | |
| Cyclofenil | Sexovid | Female infertility; Menopausal symptoms | 1970 | Mostly discontinued |
| Lasofoxifene | Fablyn | Osteoporosis prevention, treatment; Vaginal atrophy | 2009 | Only in Lithuania and Portugal |
| Ormeloxifene | Saheli | Hormonal contraception | 1991 | Only in India |
| Ospemifene | Osphena | Dyspareunia due to vaginal atrophy | 2013 | |
| Raloxifene | Evista | Osteoporosis prevention, treatment; Breast cancer prevention | 1997 | |
| Tamoxifen | Nolvadex | Breast cancer treatment | 1978 | |
| Toremifene | Fareston | Breast cancer treatment | 1997 | |
| Sources: See individual articles. | ||||
Pharmacology
Pharmacodynamics
SERMs are competitive partial agonists of the ER. Different tissues have different degrees of sensitivity to the activity of endogenous estrogens, so SERMs produce estrogenic or antiestrogenic effects depending on the specific tissue in question as well as the percentage of intrinsic activity (IA) of the SERM. An example of a SERM with high IA and thus mostly estrogenic effects is chlorotrianisene, while an example of a SERM with low IA and thus mostly antiestrogenic effects is ethamoxytriphetol. SERMs like clomifene and tamoxifen are comparatively more in the middle in their IA and their balance of estrogenic and antiestrogenic activity. Raloxifene is a SERM that is more antiestrogenic than tamoxifen; both are estrogenic in bone, but raloxifene is antiestrogenic in the uterus while tamoxifen is estrogenic in this part of the body.
| Medication | Breast | Bone | Liver | Uterus | Vagina | Brain | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipids | Coagulation | SHBG | IGF-1 | Hot flashes | Gonadotropins | |||||||||
| Estradiol | + | + | + | + | + | + | + | + | + | + | ||||
| "Ideal SERM" | – | + | + | ± | ± | ± | – | + | + | ± | ||||
| Bazedoxifene | – | + | + | + | + | ? | – | ± | – | ? | ||||
| Clomifene | – | + | + | ? | + | + | – | ? | – | ± | ||||
| Lasofoxifene | – | + | + | + | ? | ? | ± | ± | – | ? | ||||
| Ospemifene | – | + | + | + | + | + | ± | ± | – | ± | ||||
| Raloxifene | – | + | + | + | + | + | ± | – | – | ± | ||||
| Tamoxifen | – | + | + | + | + | + | + | – | – | ± | ||||
| Toremifene | – | + | + | + | + | + | + | – | – | ± | ||||
| Effect: + = Estrogenic / agonistic. ± = Mixed or neutral. – = Antiestrogenic / antagonistic. Note: SERMs generally increase gonadotropin levels in hypogonadal and eugonadal men as well as premenopausal women (antiestrogenic) but decrease gonadotropin levels in postmenopausal women (estrogenic). Sources: See template. | ||||||||||||||
| Ligand | Other names | Relative binding affinities (RBA, %)a | Absolute binding affinities (Ki, nM)a | Action | ||
|---|---|---|---|---|---|---|
| ERα | ERβ | ERα | ERβ | |||
| Estradiol | E2; 17β-Estradiol | 100 | 100 | 0.115 (0.04–0.24) | 0.15 (0.10–2.08) | Estrogen |
| Estrone | E1; 17-Ketoestradiol | 16.39 (0.7–60) | 6.5 (1.36–52) | 0.445 (0.3–1.01) | 1.75 (0.35–9.24) | Estrogen |
| Estriol | E3; 16α-OH-17β-E2 | 12.65 (4.03–56) | 26 (14.0–44.6) | 0.45 (0.35–1.4) | 0.7 (0.63–0.7) | Estrogen |
| Estetrol | E4; 15α,16α-Di-OH-17β-E2 | 4.0 | 3.0 | 4.9 | 19 | Estrogen |
| Alfatradiol | 17α-Estradiol | 20.5 (7–80.1) | 8.195 (2–42) | 0.2–0.52 | 0.43–1.2 | Metabolite |
| 16-Epiestriol | 16β-Hydroxy-17β-estradiol | 7.795 (4.94–63) | 50 | ? | ? | Metabolite |
| 17-Epiestriol | 16α-Hydroxy-17α-estradiol | 55.45 (29–103) | 79–80 | ? | ? | Metabolite |
| 16,17-Epiestriol | 16β-Hydroxy-17α-estradiol | 1.0 | 13 | ? | ? | Metabolite |
| 2-Hydroxyestradiol | 2-OH-E2 | 22 (7–81) | 11–35 | 2.5 | 1.3 | Metabolite |
| 2-Methoxyestradiol | 2-MeO-E2 | 0.0027–2.0 | 1.0 | ? | ? | Metabolite |
| 4-Hydroxyestradiol | 4-OH-E2 | 13 (8–70) | 7–56 | 1.0 | 1.9 | Metabolite |
| 4-Methoxyestradiol | 4-MeO-E2 | 2.0 | 1.0 | ? | ? | Metabolite |
| 2-Hydroxyestrone | 2-OH-E1 | 2.0–4.0 | 0.2–0.4 | ? | ? | Metabolite |
| 2-Methoxyestrone | 2-MeO-E1 | <0.001–<1 | <1 | ? | ? | Metabolite |
| 4-Hydroxyestrone | 4-OH-E1 | 1.0–2.0 | 1.0 | ? | ? | Metabolite |
| 4-Methoxyestrone | 4-MeO-E1 | <1 | <1 | ? | ? | Metabolite |
| 16α-Hydroxyestrone | 16α-OH-E1; 17-Ketoestriol | 2.0–6.5 | 35 | ? | ? | Metabolite |
| 2-Hydroxyestriol | 2-OH-E3 | 2.0 | 1.0 | ? | ? | Metabolite |
| 4-Methoxyestriol | 4-MeO-E3 | 1.0 | 1.0 | ? | ? | Metabolite |
| Estradiol sulfate | E2S; Estradiol 3-sulfate | <1 | <1 | ? | ? | Metabolite |
| Estradiol disulfate | Estradiol 3,17β-disulfate | 0.0004 | ? | ? | ? | Metabolite |
| Estradiol 3-glucuronide | E2-3G | 0.0079 | ? | ? | ? | Metabolite |
| Estradiol 17β-glucuronide | E2-17G | 0.0015 | ? | ? | ? | Metabolite |
| Estradiol 3-gluc. 17β-sulfate | E2-3G-17S | 0.0001 | ? | ? | ? | Metabolite |
| Estrone sulfate | E1S; Estrone 3-sulfate | <1 | <1 | >10 | >10 | Metabolite |
| Estradiol benzoate | EB; Estradiol 3-benzoate | 10 | ? | ? | ? | Estrogen |
| Estradiol 17β-benzoate | E2-17B | 11.3 | 32.6 | ? | ? | Estrogen |
| Estrone methyl ether | Estrone 3-methyl ether | 0.145 | ? | ? | ? | Estrogen |
| ent-Estradiol | 1-Estradiol | 1.31–12.34 | 9.44–80.07 | ? | ? | Estrogen |
| Equilin | 7-Dehydroestrone | 13 (4.0–28.9) | 13.0–49 | 0.79 | 0.36 | Estrogen |
| Equilenin | 6,8-Didehydroestrone | 2.0–15 | 7.0–20 | 0.64 | 0.62 | Estrogen |
| 17β-Dihydroequilin | 7-Dehydro-17β-estradiol | 7.9–113 | 7.9–108 | 0.09 | 0.17 | Estrogen |
| 17α-Dihydroequilin | 7-Dehydro-17α-estradiol | 18.6 (18–41) | 14–32 | 0.24 | 0.57 | Estrogen |
| 17β-Dihydroequilenin | 6,8-Didehydro-17β-estradiol | 35–68 | 90–100 | 0.15 | 0.20 | Estrogen |
| 17α-Dihydroequilenin | 6,8-Didehydro-17α-estradiol | 20 | 49 | 0.50 | 0.37 | Estrogen |
| Δ8-Estradiol | 8,9-Dehydro-17β-estradiol | 68 | 72 | 0.15 | 0.25 | Estrogen |
| Δ8-Estrone | 8,9-Dehydroestrone | 19 | 32 | 0.52 | 0.57 | Estrogen |
| Ethinylestradiol | EE; 17α-Ethynyl-17β-E2 | 120.9 (68.8–480) | 44.4 (2.0–144) | 0.02–0.05 | 0.29–0.81 | Estrogen |
| Mestranol | EE 3-methyl ether | ? | 2.5 | ? | ? | Estrogen |
| Moxestrol | RU-2858; 11β-Methoxy-EE | 35–43 | 5–20 | 0.5 | 2.6 | Estrogen |
| Methylestradiol | 17α-Methyl-17β-estradiol | 70 | 44 | ? | ? | Estrogen |
| Diethylstilbestrol | DES; Stilbestrol | 129.5 (89.1–468) | 219.63 (61.2–295) | 0.04 | 0.05 | Estrogen |
| Hexestrol | Dihydrodiethylstilbestrol | 153.6 (31–302) | 60–234 | 0.06 | 0.06 | Estrogen |
| Dienestrol | Dehydrostilbestrol | 37 (20.4–223) | 56–404 | 0.05 | 0.03 | Estrogen |
| Benzestrol (B2) | – | 114 | ? | ? | ? | Estrogen |
| Chlorotrianisene | TACE | 1.74 | ? | 15.30 | ? | Estrogen |
| Triphenylethylene | TPE | 0.074 | ? | ? | ? | Estrogen |
| Triphenylbromoethylene | TPBE | 2.69 | ? | ? | ? | Estrogen |
| Tamoxifen | ICI-46,474 | 3 (0.1–47) | 3.33 (0.28–6) | 3.4–9.69 | 2.5 | SERM |
| Afimoxifene | 4-Hydroxytamoxifen; 4-OHT | 100.1 (1.7–257) | 10 (0.98–339) | 2.3 (0.1–3.61) | 0.04–4.8 | SERM |
| Toremifene | 4-Chlorotamoxifen; 4-CT | ? | ? | 7.14–20.3 | 15.4 | SERM |
| Clomifene | MRL-41 | 25 (19.2–37.2) | 12 | 0.9 | 1.2 | SERM |
| Cyclofenil | F-6066; Sexovid | 151–152 | 243 | ? | ? | SERM |
| Nafoxidine | U-11,000A | 30.9–44 | 16 | 0.3 | 0.8 | SERM |
| Raloxifene | – | 41.2 (7.8–69) | 5.34 (0.54–16) | 0.188–0.52 | 20.2 | SERM |
| Arzoxifene | LY-353,381 | ? | ? | 0.179 | ? | SERM |
| Lasofoxifene | CP-336,156 | 10.2–166 | 19.0 | 0.229 | ? | SERM |
| Ormeloxifene | Centchroman | ? | ? | 0.313 | ? | SERM |
| Levormeloxifene | 6720-CDRI; NNC-460,020 | 1.55 | 1.88 | ? | ? | SERM |
| Ospemifene | Deaminohydroxytoremifene | 0.82–2.63 | 0.59–1.22 | ? | ? | SERM |
| Bazedoxifene | – | ? | ? | 0.053 | ? | SERM |
| Etacstil | GW-5638 | 4.30 | 11.5 | ? | ? | SERM |
| ICI-164,384 | – | 63.5 (3.70–97.7) | 166 | 0.2 | 0.08 | Antiestrogen |
| Fulvestrant | ICI-182,780 | 43.5 (9.4–325) | 21.65 (2.05–40.5) | 0.42 | 1.3 | Antiestrogen |
| Propylpyrazoletriol | PPT | 49 (10.0–89.1) | 0.12 | 0.40 | 92.8 | ERα agonist |
| 16α-LE2 | 16α-Lactone-17β-estradiol | 14.6–57 | 0.089 | 0.27 | 131 | ERα agonist |
| 16α-Iodo-E2 | 16α-Iodo-17β-estradiol | 30.2 | 2.30 | ? | ? | ERα agonist |
| Methylpiperidinopyrazole | MPP | 11 | 0.05 | ? | ? | ERα antagonist |
| Diarylpropionitrile | DPN | 0.12–0.25 | 6.6–18 | 32.4 | 1.7 | ERβ agonist |
| 8β-VE2 | 8β-Vinyl-17β-estradiol | 0.35 | 22.0–83 | 12.9 | 0.50 | ERβ agonist |
| Prinaberel | ERB-041; WAY-202,041 | 0.27 | 67–72 | ? | ? | ERβ agonist |
| ERB-196 | WAY-202,196 | ? | 180 | ? | ? | ERβ agonist |
| Erteberel | SERBA-1; LY-500,307 | ? | ? | 2.68 | 0.19 | ERβ agonist |
| SERBA-2 | – | ? | ? | 14.5 | 1.54 | ERβ agonist |
| Coumestrol | – | 9.225 (0.0117–94) | 64.125 (0.41–185) | 0.14–80.0 | 0.07–27.0 | Xenoestrogen |
| Genistein | – | 0.445 (0.0012–16) | 33.42 (0.86–87) | 2.6–126 | 0.3–12.8 | Xenoestrogen |
| Equol | – | 0.2–0.287 | 0.85 (0.10–2.85) | ? | ? | Xenoestrogen |
| Daidzein | – | 0.07 (0.0018–9.3) | 0.7865 (0.04–17.1) | 2.0 | 85.3 | Xenoestrogen |
| Biochanin A | – | 0.04 (0.022–0.15) | 0.6225 (0.010–1.2) | 174 | 8.9 | Xenoestrogen |
| Kaempferol | – | 0.07 (0.029–0.10) | 2.2 (0.002–3.00) | ? | ? | Xenoestrogen |
| Naringenin | – | 0.0054 (<0.001–0.01) | 0.15 (0.11–0.33) | ? | ? | Xenoestrogen |
| 8-Prenylnaringenin | 8-PN | 4.4 | ? | ? | ? | Xenoestrogen |
| Quercetin | – | <0.001–0.01 | 0.002–0.040 | ? | ? | Xenoestrogen |
| Ipriflavone | – | <0.01 | <0.01 | ? | ? | Xenoestrogen |
| Miroestrol | – | 0.39 | ? | ? | ? | Xenoestrogen |
| Deoxymiroestrol | – | 2.0 | ? | ? | ? | Xenoestrogen |
| β-Sitosterol | – | <0.001–0.0875 | <0.001–0.016 | ? | ? | Xenoestrogen |
| Resveratrol | – | <0.001–0.0032 | ? | ? | ? | Xenoestrogen |
| α-Zearalenol | – | 48 (13–52.5) | ? | ? | ? | Xenoestrogen |
| β-Zearalenol | – | 0.6 (0.032–13) | ? | ? | ? | Xenoestrogen |
| Zeranol | α-Zearalanol | 48–111 | ? | ? | ? | Xenoestrogen |
| Taleranol | β-Zearalanol | 16 (13–17.8) | 14 | 0.8 | 0.9 | Xenoestrogen |
| Zearalenone | ZEN | 7.68 (2.04–28) | 9.45 (2.43–31.5) | ? | ? | Xenoestrogen |
| Zearalanone | ZAN | 0.51 | ? | ? | ? | Xenoestrogen |
| Bisphenol A | BPA | 0.0315 (0.008–1.0) | 0.135 (0.002–4.23) | 195 | 35 | Xenoestrogen |
| Endosulfan | EDS | <0.001–<0.01 | <0.01 | ? | ? | Xenoestrogen |
| Kepone | Chlordecone | 0.0069–0.2 | ? | ? | ? | Xenoestrogen |
| o,p'-DDT | – | 0.0073–0.4 | ? | ? | ? | Xenoestrogen |
| p,p'-DDT | – | 0.03 | ? | ? | ? | Xenoestrogen |
| Methoxychlor | p,p'-Dimethoxy-DDT | 0.01 (<0.001–0.02) | 0.01–0.13 | ? | ? | Xenoestrogen |
| HPTE | Hydroxychlor; p,p'-OH-DDT | 1.2–1.7 | ? | ? | ? | Xenoestrogen |
| Testosterone | T; 4-Androstenolone | <0.0001–<0.01 | <0.002–0.040 | >5000 | >5000 | Androgen |
| Dihydrotestosterone | DHT; 5α-Androstanolone | 0.01 (<0.001–0.05) | 0.0059–0.17 | 221–>5000 | 73–1688 | Androgen |
| Nandrolone | 19-Nortestosterone; 19-NT | 0.01 | 0.23 | 765 | 53 | Androgen |
| Dehydroepiandrosterone | DHEA; Prasterone | 0.038 (<0.001–0.04) | 0.019–0.07 | 245–1053 | 163–515 | Androgen |
| 5-Androstenediol | A5; Androstenediol | 6 | 17 | 3.6 | 0.9 | Androgen |
| 4-Androstenediol | – | 0.5 | 0.6 | 23 | 19 | Androgen |
| 4-Androstenedione | A4; Androstenedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| 3α-Androstanediol | 3α-Adiol | 0.07 | 0.3 | 260 | 48 | Androgen |
| 3β-Androstanediol | 3β-Adiol | 3 | 7 | 6 | 2 | Androgen |
| Androstanedione | 5α-Androstanedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| Etiocholanedione | 5β-Androstanedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| Methyltestosterone | 17α-Methyltestosterone | <0.0001 | ? | ? | ? | Androgen |
| Ethinyl-3α-androstanediol | 17α-Ethynyl-3α-adiol | 4.0 | <0.07 | ? | ? | Estrogen |
| Ethinyl-3β-androstanediol | 17α-Ethynyl-3β-adiol | 50 | 5.6 | ? | ? | Estrogen |
| Progesterone | P4; 4-Pregnenedione | <0.001–0.6 | <0.001–0.010 | ? | ? | Progestogen |
| Norethisterone | NET; 17α-Ethynyl-19-NT | 0.085 (0.0015–<0.1) | 0.1 (0.01–0.3) | 152 | 1084 | Progestogen |
| Norethynodrel | 5(10)-Norethisterone | 0.5 (0.3–0.7) | <0.1–0.22 | 14 | 53 | Progestogen |
| Tibolone | 7α-Methylnorethynodrel | 0.5 (0.45–2.0) | 0.2–0.076 | ? | ? | Progestogen |
| Δ4-Tibolone | 7α-Methylnorethisterone | 0.069–<0.1 | 0.027–<0.1 | ? | ? | Progestogen |
| 3α-Hydroxytibolone | – | 2.5 (1.06–5.0) | 0.6–0.8 | ? | ? | Progestogen |
| 3β-Hydroxytibolone | – | 1.6 (0.75–1.9) | 0.070–0.1 | ? | ? | Progestogen |
| Footnotes: a = (1) Binding affinity values are of the format "median (range)" (# (#–#)), "range" (#–#), or "value" (#) depending on the values available. The full sets of values within the ranges can be found in the Wiki code. (2) Binding affinities were determined via displacement studies in a variety of in-vitro systems with labeled estradiol and human ERα and ERβ proteins (except the ERβ values from Kuiper et al. (1997), which are rat ERβ). Sources: See template page. | ||||||
Binding site
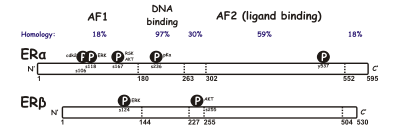
SERM act on the estrogen receptor (ER), which is an intracellular, ligand-dependent transcriptional activator and belongs to the nuclear receptor family. Two different subtypes of ER have been identified, ERα and ERβ. ERα is considered the main medium where estrogen signals are transduced at the transcriptional level and is the predominant ER in the female reproductive tract and mammary glands while ERβ is primarily in vascular endothelial cells, bone, and male prostate tissue. ERα and ERβ concentration are known to be different in tissues during development, aging or disease state. Many characteristics are similar between these two types such as size (~600 and 530 amino acids) and structure. ERα and ERβ share approximately 97% of the amino-acid sequence identity in the DNA-binding domain and about 56% in the ligand-binding domain. The main difference of the ligand-binding domains is determined by Leu-384 and Met-421 in ERα, which are replaced by Met-336 and Ile-373, respectively, in ERβ. The variation is greater on the N-terminus between ERα and ERβ.
DNA-binding domain consists of two subdomains. One with a proximal box that is involved in DNA recognition while the other contains a distal box responsible for DNA-dependent, DNA-binding domain dimerization. The proximal box sequence is identical between ERα and ERβ, which indicates similar specificity and affinity between the two subgroups. DNA-binding domain's globular proteins contain eight cysteines and allow for a tetrahedral coordination of two zinc ions. This coordination makes the binding of ER to estrogen response elements possible. Ligand-binding domain is a globular, three-layered structure made of 11 helixes and contains a pocket for the natural or synthetic ligand. Influencing factors for binding affinity are mainly the presence of a phenol moiety, molecular size and shape, double bonds and hydrophobicity.
The differential positioning of the activating function 2 (AF-2) helix 12 in the ligand-binding domain by the bound ligand determines whether the ligand has an agonistic and antagonistic effect. In agonist-bound receptors, helix 12 is positioned adjacent to helices 3 and 5. Helices 3, 5, and 12 together form a binding surface for an NR box motif contained in coactivators with the canonical sequence LXXLL (where L represents leucine or isoleucine and X is any amino acid). Unliganded (apo) receptors or receptors bound to antagonist ligands turn helix 12 away from the LXXLL-binding surface that leads to preferential binding of a longer leucine-rich motif, LXXXIXXX(I/L), present on the corepressors NCoR1 or SMRT. In addition, some cofactors bind to ER through the terminals, the DNA-binding site or other binding sites. Thus, one compound can be an ER agonist in a tissue rich in coactivators but an ER antagonist in tissues rich in corepressors.
Mechanism of action
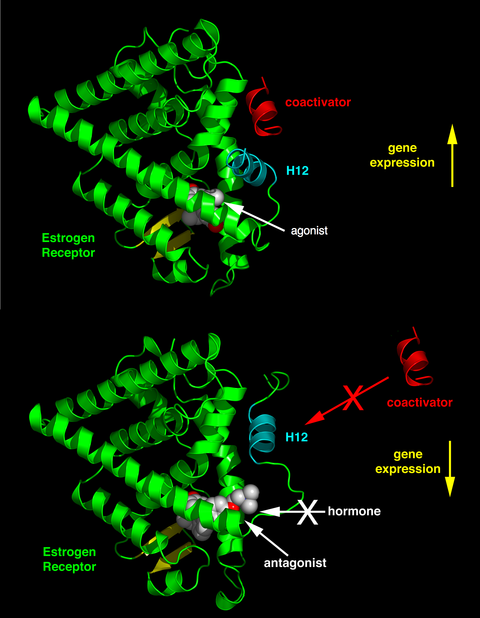
Estrogenic compounds span a spectrum of activity ranging from:
- Full agonists (agonistic in all tissues) such as the natural endogenous hormone estradiol
- Mixed agonists/antagonistics (agonistic in some tissues while antagonistic in others) such as tamoxifen (a SERM).
- Pure antagonists (antagonistic in all tissues) such as fulvestrant.
SERMs are known to stimulate estrogenic actions in tissues such as the liver, bone and cardiovascular system but known to block estrogen action where stimulation is not desirable, such as in the breast and the uterus. This agonistic or antagonistic activity causes varied structural changes of the receptors, which results in activation or repression of the estrogen target genes. SERMs interact with receptors by diffusing into cells and there binding to ERα or ERβ subunits, which results in dimerization and structural changes of the receptors. This makes it easier for the SERMs to interact with estrogen response elements which leads to the activation of estrogen-inducible genes and mediating the estrogen effects.
SERMs unique feature is their tissue- and cell-selective activity. There is growing evidence to support that SERM activity is mainly determined by selective recruitment of corepressors and coactivators to ER target genes in specific types of tissues and cells. SERMs can impact coactivator protein stability and can also regulate coactivator activity through post-translational modifications such as phosphorylation. Multiple growth signaling pathways, such as HER2, PKC, PI3K and more, are downregulated in response to anti-estrogen treatment. Steroid receptor coactivator 3 (SRC-3) is phosphorylated by activated kinases that also enhance its coactivator activity, affect cell growth and ultimately contribute to drug resistance.
The ratio of ERα and ERβ at a target site may be another way SERM activity is determined. High levels of cellular proliferation correlate well with a high ERα:ERβ ratio, but repression of cellular proliferation correlates to ERβ being dominant over ERα. The ratio of ERs in neoplastic and normal breast tissue could be important when considering chemoprevention with SERMs.
When looking at the differences between ERα and ERβ, Activating Function 1 (AF-1) and AF-2 are important. Together they play an important part in the interaction with other co-regulatory proteins that control gene transcription. AF-1 is located in the amino terminus of the ER and is only 20% homologous in ERα and ERβ. On the other hand, AF-2 is very similar in ERα and ERβ, and only one amino acid is different. Studies have shown that by switching AF-1 regions in ERα and ERβ, that there are specific differences in transcription activity. Generally, SERMs can partially activate engineered genes through ERα by an estrogen receptor element, but not through ERβ. Although, raloxifene and the active form of tamoxifen can stimulate AF-1-regulated reporter genes in both ERα and ERβ.
Because of the discovery that there are two ER subtypes, it has brought about the synthesis of a range of receptor specific ligands that can switch on or off a particular receptor. However, the external shape of the resulting complex is what becomes the catalyst for changing the response at a tissue target to a SERM.
X-ray crystallography of estrogens or antiestrogens has shown how ligands program the receptor complex to interact with other proteins. The ligand-binding domain of the ER demonstrates how ligands promote and prevent coactivator binding based on the shape of the estrogen or antiestrogen complex. The broad range of ligands that bind to the ER can create a spectrum of ER complexes that are fully estrogenic or antiestrogenic at a specific target site. The main result of a ligand-binding to ER is a structural rearrangement of the ligand-binding pocket, primarily in the AF-2 of the C-terminal region. The binding of ligands to ER leads to the formation of a hydrophobic pocket that regulates cofactors and receptor pharmacology. The correct folding of ligand-binding domain is required for activation of transcription and for ER to interact with a number of coactivators.
Coactivators are not just protein partners that connect sites together in a complex. Coactivators play an active role in modifying the activity of a complex. Post-translation modification of coactivators can result in a dynamic model of steroid hormone action by way of multiple kinase pathways initiated by cell surface growth factor receptors. Under the guidance of a multitude of protein remodelers to form a multiprotein coactivator complex that can interact with the phosphorylated ER at a specific gene promoter site, the core coactivator first has to recruit a specific set of cocoactivators. The proteins that the core coactivator assembles as the core coactivated complex have individual enzymatic activities to methylate or acetylate adjacent proteins. The ER substrates or coenzyme A can be polyubiquitinated by multiple cycles of the reaction or, depending on linkage proteins, they can either be activated further or degraded by the 26S proteasome.
Consequently, to have an effective gene transcription that is programmed and targeted by the structure and phosphorylation status of the ER and coactivators, it is required to have a dynamic and cyclic process of remodeling capacity for transcriptional assembly, after which the transcription complex is then instantly routinely destroyed by the proteasome.
Structure and function
Structure–activity relationships
The core structure of SERMs simulates the 17β-estradiol template. They have two aromatic rings separated by 1-3 atoms (often a stilbene-type of arrangement). Between the two phenyls of the core, SERMs typically have a 4-substituted phenyl group that, when bound to ER, projects from a position of an estratriene nucleus so that helix 12 moves from the receptor opening and blocks the space where coactivator proteins would normally bind and cause ER agonist activity. There has been a lot of variations in the core portion of SERMs while there has been less flexibility with what is tolerated in the side chain. SERMs can be classified by their core structure.
First-generation triphenylethylenes

The first main structural class of SERM-type molecules reported are the triphenylethylenes. The stilbene core (similar to the nonsteroidal estrogen, diethylstilbestrol) essentially mimics steroidal estrogens such as 17β-estradiol, while the side chain overlays with the 11th position of the steroid nucleus. Triphenylethylene derivatives have an additional phenyl group attached to the ethylene bridge group. The 3-position H-bonding ability of phenols is a significant requirement for ER binding.

The first drug, clomifene, has a chloro-substituent on the ethylene side chain which produces similar binding affinities as the later discovered drug tamoxifen. Clomifene is a mixture of estrogenic (cis-form) and antiestrogenic isomers (trans-form). Cis and trans are defined in terms of the geometric relationships of the two unsubstituted phenyl rings. The two isomers of clomifene have different profiles, where the trans-form has activity more similar to tamoxifen while the cis-form behaves more like 17β-estradiol. Cis is approximately ten times more potent than trans. However, trans isomer is the most potent stimulator of epithelial cell hypertrophy since clomifene is antagonistic at low doses and agonistic at high doses. The antagonist isomers may cause inhibitory estrogenic effects in the uterus and mammary cancers, but the estrogenic isomer could combine with novel receptors to produce estrogen-like effects in bone.

Tamoxifen has become the treatment of choice for women diagnosed with all stages of hormone-responsive breast cancer, that is, breast cancer that is both ER and/or progesterone positive. In the US, it is also administered for prophylactic chemoprevention in women identified as high risk for breast cancer. Tamoxifen is a pure antiestrogenic trans-isomer and has differential actions at estrogen target tissues throughout the body. Tamoxifen is selectively antiestrogenic in the breast but estrogen-like in bones and endometrial cancer. Tamoxifen undergo phase I metabolism in the liver by microsomal cytochrome P450 (CYP) enzymes. The major metabolites of tamoxifen are N-desmethyltamoxifen and 4-hydroxytamoxifen.
The crystallographic structure of 4-hydroxytamoxifen interacts with the amino acids of the ER within the ligand-binding domain. The contact between the phenolic group, water molecule, and glutamate and arginine in the receptor (ERα; Glu 353/Arg 394) resolves in high affinity binding so that 4-hydroxy tamoxifen, with a phenolic ring that resembles the A ring of 17β-estradiol, has more than 100 times higher relative binding affinity than tamoxifen, which has no phenol. If its OH group is eliminated or its position is changed the binding affinity is reduced.
The triphenylethylene moiety and the side chain are required for tamoxifen binding to the ER, whereas for 4-hydroxytamoxifen, the side chain, and the phenyl-propene do not appear as crucial structural elements for binding to the ER. The basicity and length of the side chain do not seem to play a crucial role for tamoxifen binding affinity to the ER nor the β-ring of tamoxifen, but the stilbene moiety of tamoxifen is necessary for binding to the ER. The hydroxyl group is of particular importance for ER binding of 4-hydroxytamoxifen, and the ethyl side chain of tamoxifen protrudes out of the ligand-binding domain of the ER.
Few tamoxifen users have had increased rates of uterine cancer, hot flushes, and thromboembolisms. The drug can also cause hepatocarcinomas in rats. This is likely due to the ethyl group of the tamoxifen stilbene core that is subject to allylic oxidative activation causing DNA alkylation and strand scission. This problem is later corrected in toremifene. Tamoxifen is more promiscuous than raloxifene in target sites because of the relationship between ER's amino acid in Asp-351 and the antiestrogenic side chain of the SERM. The side chain for tamoxifen cannot neutralize Asp-351, so the site allosterically influences AF-1 at the proximal end of the ER. This issue is mended with the second-generation drug raloxifene.

Toremifene is a chlorinated derivative of the nonsteroidal triphenylethylene antiestrogen tamoxifen with a chloro substituent at the ethylene side chain producing similar binding affinities to that of tamoxifen. The structure and activity relationship of toremifene is similar to that of tamoxifen, but it has a substantial improvement from the older drug in regards to DNA alkylation. The presence of the added chlorine atom reduces the stability of cations formed from activated allylic metabolites and thus decreases alkylation potential, and indeed toremifene does not display DNA adduct formation in rodent hepatocytes. Toremifene protects against bone loss in ovariectomized rat models and affects bone resorption markers clinically in a similar fashion to tamoxifen. Toremifene undergoes phase I metabolism by microsomal cytochrome P450 enzymes, like tamoxifen, but primarily by the CYP3A4 isoform. Toremifene forms its two major metabolites N-desmethyltoremifene and deaminohydroxy-toremifene (ospemifene) by undergoing N-demethylation and deamination-hydroxylation. N-desmethyltoremifene has similar efficacy as toremifene while 4-hydroxytoremifene has a higher binding affinity to the ER than toremifene. 4-hydroxytoremifene has a role similar to that of 4-hydroxytamoxifen.
Second-generation benzothiophenes

Raloxifene belongs to the second-generation benzothiophene SERM drugs. It has a high affinity for the ER with potent antiestrogenic activity and tissue-specific effects distinct from estradiol. Raloxifene is an ER agonist in bone and the cardiovascular system, but in breast tissue and the endometrium it acts as an ER antagonist. It is extensively metabolized by glucuronide conjugation in the gut and because of that has a low bioavailability of only 2% while that of tamoxifen and toremifene is approximately 100%.
The advantage of raloxifene over the triphenylethylene tamoxifen is reduced effect on the uterus. The flexible hinge group, as well as the antiestrogenic phenyl 4-piperidinoethoxy side chain, are important for minimizing uterine effects. Because of its flexibility the side chain can obtain an orthogonal disposition relative to the core so that the amine of raloxifens side chain is 1 Å closer than tamoxifens to amino acid Asp-351 in ERα's ligand-binding domain.
The critical role of the intimate relationship between the hydrophobic side chain of raloxifene and the hydrophobic residue of the receptor to change both the shape and charge of the external surface of a SERM-ER complex has been confirmed with raloxifene derivatives. When the interactive distance between raloxifene and Asp-351 is increased from 2.7 Å to 3.5-5 Å it causes increased estrogen-like action of the raloxifene-ERα complex. When the piperidine ring of raloxifene is replaced by cyclohexane, the ligand loses antiestrogenic properties and becomes a full agonist. The interaction between SERM's antiestrogenic side chain and amino acid Asp-351 is the important first step in silencing AF-2. It relocates helix 12 away from the ligand-binding pocket thereby preventing coactivators from binding to the SERM-ER complex.
Third-generation

Third-generation compounds display either no uterine stimulation, improved potency, no significant increases in hot flushes or even a combination of these positive attributes.
The first dihydronapthalene SERM, nafoxidine, was a clinical candidate for the treatment of breast cancer but had side effects including severe phototoxicity. Nafoxidine has all three phenyls constrained in a coplanar arrangement like tamoxifen. But with hydrogenation, the double bond of nafoxidene were reduced, and both phenyls are cis-oriented. The amine-bearing side chain can then adopt an axial conformation and locate this group orthogonally to the plane of the core, like ralofoxifene and other less uterotropic SERMs.

Modifications of nafoxidine resulted in lasofoxifene. Lasofoxifene is among the most potent SERMs reported in protection against bone loss and cholesterol reduction. The excellent oral potency of lasofoxifene has been attributed to reduced intestinal glucuronidation of the phenol. Unlike raloxifene, lasofoxifene satisfies the requirement of a pharmacophore model that predicts resistance to gut wall glucuronidation. The structural requirement is a non-planar topology with the steric bulk close to the plane of a fused bicyclic aromatic system. The interactions between the ER and lasofoxifene are consistent with the general features of SERM-ER recognition. Lasofoxifene's large flexible side chain terminates in a pyrrolidine head group and threads its way out toward the surface of the protein, where it interferes directly with the positioning of the AF-2 helix. A salt bridge forms between lasofoxifene and Asp-351. The charge neutralization in this region ER may explain some antiestrogenic effects exerted by lasofoxifene.

The indole system has served as a core unit in SERMs, and when an amine is attached to the indole with a benzyloxyethyl, the resultant compounds were shown to have no preclinical uterine activity while sparing rat bone with full efficacy at low doses. Bazedoxifene is one of those compounds. The core binding domain consists of a 2-phenyl-3-methyl indole and a hexamethylenamine ring at the side chain affecter region. It is metabolized by glucuronidation, with the absolute bioavailability of 6.2%, 3-fold higher than that of raloxifene. It has agonistic effects on bone and lipid metabolism but not on breast and uterine endometrium. It is well tolerated and displays no increase in hot flush incidences, uterine hypertrophy or breast tenderness.

Ospemifene is a triphenylethylene and a known metabolite of toremifene. It's structurally very similar to tamoxifen and toremifene. Ospemifene does not have 2-(dimethylamino)ethoxy group as tamoxifen. Structure–activity relationship studies showed that by removing that group of tamoxifen agonistic activity in the uterus was significantly reduced, but not in bone and cardiovascular system. Preclinical and clinical data show that ospemifene is well tolerated with no major side effects. Benefits that ospemifene may have over other SERMs is its neutral effect on hot flushes and ER-agonist effect on the vagina, improving the symptoms of vaginal dryness.
Binding modes

The SERMs are known to feature four distinctive modes of binding to ER. One of those features are strong hydrogen bonds between the ligand and ERα's Arg-394 and Glu-353 that line the "A-ring pocket" and help the ligand to stay in ER's binding pocket. This is unlike 17β-estradiol which is hydrogen bonded to His-524 in the "D-ring pocket". Other distinctive bindings to the ligand-binding pocket are with a nearly planar "core" structure typically composed of a biaryl heterocycle, equivalent to the A-ring and B-ring of 17β-estradiol, to the corresponding binding site; a bulky side chain from the biaryl structure, analogous to the B-ring of 17β-estradiol and finally a second side group that is the C- and D-ring equivalent and usually aromatic, fills the remainder volume of the ligand-binding pocket.
The small differences between the two subtypes of ER have been used to develop subtype-selective ER modulators, but the high similarity between the two receptors make the development very challenging. Amino acids in the ligand-binding domains differ at two positions, Leu-384 and Met-421 in ERα and Met-336 and Ile-373 in ERβ, but they have similar hydrophobicity and occupying volumes. However, the shapes and the rotational barrier of the amino acid residues are not the same, leading to distinguish α- and β-face of the binding cavity between ERα and ERβ. This causes ERα-preferential-binding of ligand substituents that are aligned downwards facing Met-336 while ligand substituents aligned upwards facing Met-336 are more likely to bind to ERβ. Another difference is in Val-392 in ERα, which is replaced by Met-344 in ERβ. ERβ's binding pocket volume is slightly smaller and the shape a bit different from ERα's. Many ERβ-selective ligands have a largely planar arrangement as the binding cavity of ERβ is slightly narrower than that of ERα, however, this by itself leads to modest selectivity. To attain strong selectivity, the ligand must place substituents very close to one or more of the amino acid differences between ERα and ERβ in order to create a strong repulsive force towards the other subtype receptor. In addition, the structure of the ligand must be rigid. Repulsive interactions may otherwise lead to conformational change of the ligand and, therefore, creating alternative binding modes.
First-generation triphenylethylenes
Tamoxifen is converted by the liver cytochrome P450 into the 4-hydroxytamoxifen and is a more selective antagonist of the ERα subtype than ERβ. 4-hydroxytamoxifen binds to ERs within the same binding pocket that recognizes 17β-estradiol. The receptor recognition of 4-hydroxytamoxifen appears to be controlled by two structural features of 4-hydroxytamoxifen, the phenolic A ring, and the bulky side chain. The phenolic A ring forms hydrogen bonds to the side groups of ER's Arg-394, Glu-354 and to structurally conserved water. The bulky side chain, protruding from the binding cavity, displaces helix 12 from ligand-binding pocket to cover part of the coactivator binding pocket. The ER-4-hydroxytamoxifen complex formation recruits corepressors proteins. This leads to decreased DNA synthesis and inhibition of estrogen activity. Clomifene and torimefene produce binding affinities similar to that of tamoxifen. Thus, these two drugs are more selective antagonists of the ERα subtype than ERβ.
Second-generation benzothiophenes

Raloxifene, like 4-hydroxytamoxifen, binds to ERα with the hydroxyl group of its phenolic "A ring" through hydrogen bonds with Arg-394 and Glu-353. In addition to these bonds, raloxifene forms a second hydrogen bond to ER through the side group of His-524 because of the presence of a second hydroxyl group in the "D ring". This hydrogen bond is also unlike that between 17β-estradiol and His-524, as the imidazole ring of His-524 is rotated to counteract the difference of the oxygen position in raloxifene and in 17β-estradiol. Just like in 4-hydroxytamoxifen, the bulky side chain of raloxifene displaces helix 12.
Third-generation
Lasofoxifene interaction with ERα is typical of those between SERM-ERα such as a nearly planar topology (the tetrahydronapthalene carbocycle), hydrogen bonding with Arg-394 and Glu-353 and the phenyl side chains of lasofoxifene filling the C-ring and D-ring volume of the ligand-binding pocket. Lasofoxifene diverts helix 12 and prevents the binding of coactivator proteins with LXXLL motives. This is achieved by lasofoxifene occupying the space normally filled by Leu-540's side group and modulating the conformation of residues of helix 11 (His-524, Leu-525). Furthermore, lasofoxifene also directly interferes with helix 12 positioning by the drug's ethyl pyrrolidine group. In vitro studies indicate that bazedoxifene competitively blocks 17β-estradiol by high and similar binding to both ERα and ERβ. Bazedoxifenes main binding domain consists of the 2-phenyl-3-methylindole and a hexamethylenamine ring at the side chain affected region.
Ospemifene is an oxidative deaminated metabolite of toremifene as has a similar binding to ER as toremifene and tamoxifen. The competitive binding to ERα and ERβ of the three metabolites 4-hydroxy Ospemifene, 4'-hydroxy Ospemifene and the 4-hydroxy-, side chain carboxylic acid Ospemifene is at least as high as the parent compound.
History

The discovery of SERMs resulted from attempts to develop new contraceptives. Clomifene and tamoxifen prevented conception in rats but did the opposite in humans. Clomifene successfully induced ovulation in subfertile women and on February 1, 1967, it was approved in the US for the treatment of ovulation dysfunction in women who were trying to conceive.Toxicological issues prevented long term use of clomifene and further drug development for other potential applications such as breast cancer treatment and prevention.
It was another ten years before tamoxifen was approved in December 1977, not as a contraceptive but as a hormonal treatment to treat and prevent breast cancer. The discovery in 1987 that the SERMs tamoxifen and raloxifene, then thought to be antiestrogens because of antagonist effects in breast tissue, showed estrogenic effects in preventing bone loss in ovariectomized rats had a great effect on our understanding of the function of estrogen receptors and nuclear receptors in general. The term SERM was introduced to describe these compounds that have a combination of estrogen agonist, partial agonist, or antagonist activities depending on the tissue. Toremifene has been shown to be compatible with tamoxifen, and in 1996 it was approved for use in the treatment of breast cancer in postmenopausal women.
Raloxifene originally failed as a breast cancer drug due to its poor performance in comparison to tamoxifen in the laboratory but the estrogenic effects of raloxifene on bone led to its rediscovery and approval in 1997. It was approved for prevention and treatment of osteoporosis and was the first clinically available SERM to prevent both osteoporosis and breast cancer.Ospemifene was approved on February 26, 2013, for the treatment of moderate to severe dyspareunia, which is a symptom, due to menopause, of vulvar and vaginal atrophy. Combined therapy with conjugated estrogens and the SERM bazedoxifene, was approved on October 3, 2013, for the treatment of vasomotor symptoms linked with menopause. Bazedoxifene is also used in the prevention of postmenopausal osteoporosis. The search for a potent SERM with bone efficacy and better bioavailability than raloxifene led to the discovery of lasofoxifene. Although lasofoxifene was approved in 2009, it was not marketed for three years following the approval, so the marketing authorization for it has expired. In Europe, bazedoxifene is indicated for the treatment of osteoporosis in postmenopausal women at increased risk of fracture while in India ormeloxifene has been used for dysfunctional uterine bleeding and birth control.
See also
- Estrogen deprivation therapy
- List of selective estrogen receptor modulators
- Selective androgen receptor modulator
- Selective estrogen receptor degrader
- Selective receptor modulator
- Timeline of cancer treatment development