
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Clomid, Serophene, others |
| Other names | Clomiphene; Chloramifene; Chloramiphene; MRL-41; MRL/41; NSC-35770 |
| AHFS/Drugs.com | Monograph |
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Selective estrogen receptor modulator; Progonadotropin |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | High (>90%) |
| Metabolism | Liver CYP2D6 (with enterohepatic circulation) |
| Metabolites | 4-Hydroxyclomiphene (4-OH-CLO), 4-Hydroxy-N-desethylclomiphene (4-OH-DE-CLO) |
| Elimination half-life | 4 – 7 days active metabolites: |
| Excretion | Mainly feces, some in urine |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.011.826 |
| Chemical and physical data | |
| Formula | C26H28ClNO |
| Molar mass | 405.966 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
|
| |
Clomifene, also known as clomiphene, is a medication used to treat infertility in women who do not ovulate, including those with polycystic ovary syndrome. Use results in a greater chance of twins. It is taken by mouth once a day, with a course of treatment that usually lasts for five days.
Common side effects include pelvic pain and hot flashes. Other side effects can include changes in vision, vomiting, trouble sleeping, ovarian cancer, and seizures. It is not recommended in people with liver disease or abnormal vaginal bleeding of unknown cause or who are pregnant. Clomifene is in the selective estrogen receptor modulator (SERM) family of medication and is a nonsteroidal medication. It works by causing the release of GnRH by the hypothalamus, and subsequently gonadotropin from the anterior pituitary.
Clomifene was approved for medical use in the United States in 1967. It is on the World Health Organization's List of Essential Medicines, under the category "Ovulation inducers" (Complementary List). Its introduction began the era of assisted reproductive technology.
Clomifene (particularly the purified enclomiphene isomer) has also been found to have a powerful ability to boost or restore testosterone levels in hypogonadal men.
Medical uses
Ovulation induction
Clomifene is one of several alternatives for ovulation induction in those who are infertile due to anovulation or oligoovulation. Evidence is lacking for the use of clomifene in those who are infertile without a known reason. In such cases, studies have observed a clinical pregnancy rate 5.6% per cycle with clomifene treatment vs. 1.3%–4.2% per cycle without treatment.
Proper timing of the drug is important; it should be taken starting on about the fifth day of the cycle, and there should be frequent intercourse.
The following procedures may be used to monitor induced cycles:
- Follicular monitoring with vaginal ultrasound, starting 4–6 days after the last pill. Serial transvaginal ultrasound can reveal the size and number of developing follicles. It can also provide presumptive evidence of ovulation such as the sudden collapse of the preovulatory follicle, and an increase in fluid volume in the rectouterine pouch. After ovulation, it may reveal signs of luteinization such as loss of clearly defined follicular margins and appearance of internal echoes.
- Serum estradiol levels, starting 4–6 days after the last pill
- Adequacy of LH surge by urine LH surge tests 3 to 4 days after last clomifene pill
- Post-coital test 1–3 days before ovulation to check whether there are at least 5 progressive sperm per HPF
- Mid-luteal progesterone, with at least 10 ng/ml 7–9 days after ovulation being regarded as adequate.
Repeat dosing: This 5-day treatment course can be repeated every 30 days. The dosage may be increased by 50-mg increments in subsequent cycles until ovulation is achieved. It is not recommended by the manufacturer to use clomifene for more than 6 cycles.
It is no longer recommended to perform an ultrasound examination to exclude any significant residual ovarian enlargement before each new treatment cycle.
Other uses
Clomifene has also been used with other assisted reproductive technology to increase success rates of these other modalities.
Clomifene is sometimes used in the treatment of male hypogonadism as an alternative to testosterone replacement therapy. The medication has been used at a dosage of 20 to 50 mg three times per week to once daily for this indication. It has been found to increase testosterone levels by 2- to 2.5-times in hypogonadal men at such dosages. Despite the use of questionnaires in testosterone replacement comparator trials being called into question, clomifene's lower cost, therapeutic benefits, and greater value towards hypogonadism improvement have been noted.
Clomifene consists of two stereoisomers in equal proportion: enclomifene and zuclomifene. Zuclomifene has pro-estrogenic properties, whereas enclomifene is pro-androgenic, i.e. it promotes testosterone production through stimulation of the HPG axis. For this reason, purified enclomifene isomer has been found to be twice as effective in boosting testosterone compared to the standard mix of both isomers. Additionally, enclomifene has a half-life of just 10 hours, but zuclomifene has a half-life on the order of several days to a week, so if the goal is to boost testosterone, taking regular clomifene may produce far longer-lasting pro-estrogenic effects than pro-androgenic effects.
Clomifene has been used in the treatment of gynecomastia. It has been found to be useful in the treatment of some cases of gynecomastia but it is not as effective as tamoxifen or raloxifene for this indication. It has shown variable results for gynecomastia (probably because the zuclomifene isomer is estrogenic), and hence is not recommended for treatment of the condition. Pure enclomifene isomer is likely to be more effective than clomifene at treating gynecomastia, because of the lack of the zuclomifene isomer (as noted above).
Due to its long half-life, zuclomifene can be detected in urine for at least 261 days after discontinuation (261 days after discontinuation with a half-life of 30 days, there is still 0.24% of the peak level of zuclomifene being excreted, whereas with a half-life of 10 hours, enclomifene reaches the same 0.24% level in less than 4 days).
Because of its potential for boosting testosterone, clomifene is listed as banned for use by competitive sportsmen, both in and out of competition, by the World Anti-Doping Agency, absent an organic etiology of primary hypogonadism.
Contraindications
Contraindications include an allergy to the medication, pregnancy, prior liver problems, abnormal vaginal bleeding of unclear cause, ovarian cysts other than those due to polycystic ovarian syndrome, unmanaged adrenal or thyroid problems, and pituitary tumors.
Side effects
The most common adverse drug reaction associated with the use of clomifene (>10% of people) is reversible ovarian enlargement.
Less common effects (1–10% of people) include visual symptoms (blurred vision, double vision, floaters, eye sensitivity to light, scotomata), headaches, vasomotor flushes (or hot flashes), light sensitivity and pupil constriction, abnormal uterine bleeding and/or abdominal discomfort.
Rare adverse events (<1% of people) include: high blood level of triglycerides, liver inflammation, reversible baldness and/or ovarian hyperstimulation syndrome.
Clomifene can lead to multiple ovulation, hence increasing the chance of twins (10% of births instead of ~1% in the general population) and triplets.
Rates of birth defects and miscarriages do not appear to change with the use of clomifene for fertility. Clomifene has been associated with liver abnormalities and a couple of cases of hepatotoxicity.
Cancer risk
Some studies have suggested that clomifene if used for more than a year may increase the risk of ovarian cancer. This may only be the case in those who have never been and do not become pregnant. Subsequent studies have failed to support those findings.
Clomifene has been shown to be associated with an increased risk of malignant melanomas and thyroid cancer. Thyroid cancer risk was not associated with the number of pregnancies carried to viability.
Pharmacology
Pharmacodynamics
Selective estrogen receptor modulator activity
Clomifene is a nonsteroidal triphenylethylene derivative that acts as a selective estrogen receptor modulator (SERM). It consists of a non-racemic mixture of zuclomifene (~38%) and enclomifene (~62%), each of which has unique pharmacologic properties. It is a mixed agonist and antagonist of the estrogen receptor (ER). Clomifene activates the ERα in the setting of low baseline estrogen levels and partially blocks the receptor in the context of high baseline estrogen levels. Conversely, it is an antagonist of the ERβ. Clomifene has antiestrogenic effects in the uterus. There is little clinical research on the influence of clomifene in many target tissues, such as lipids, the cardiovascular system, and the breasts. Positive effects of clomifene on bone have been observed. Clomifene has been found to decrease insulin-like growth factor 1 (IGF-1) levels in women.
Clomifene is a long-acting ER ligand, with a nuclear retention of greater than 48 hours. Clomifene is a prodrug being activated via similar metabolic pathways as the related triphenylethylene SERMs tamoxifen and toremifene. The affinity of clomifene for the ER relative to estradiol ranges from 0.1 to 12% in different studies, which is similar to the range for tamoxifen (0.06–16%). 4-Hydroxyclomifene, a major active metabolite of clomifene, and afimoxifene (4-hydroxytamoxifen), a major active metabolite of tamoxifen, show 89–251% and 41–246% of the affinity of estradiol for the ER in human MCF-7 breast cancer cells, respectively. The ER affinities of the isomers of 4-hydroxyclomifene were 285% for (E)-4-hydroxyclomifene and 16% for (Z)-4-hydroxyclomifene relative to estradiol. 4-Hydroxy-N-desmethylclomifene has similar affinity to 4-hydroxyclomifene for the ER. In one study, the affinities of clomifene and its metabolites for the ERα were ~100 nM for clomifene, ~2.4 nM for 4-hydroxyclomifene, ~125 nM for N-desmethylclomifene, and ~1.4 nM for 4-hydroxy-N-desmethylclomifene.
Even though clomifene has some estrogenic effect, the antiestrogenic property is believed to be the primary source for stimulating ovulation. Clomifene appears to act mostly in the hypothalamus where it depletes hypothalamic ERs and blocks the negative feedback effect of circulating endogenous estradiol, which in turn results in an increase in hypothalamic gonadotropin-releasing hormone (GnRH) pulse frequency and circulating concentrations of follicle-stimulating hormone (FSH) and luteinizing hormone (LH).
In normal physiologic female hormonal cycling, at 7 days past ovulation, high levels of estrogen and progesterone produced from the corpus luteum inhibit GnRH, FSH, and LH at the hypothalamus and anterior pituitary. If fertilization does not occur in the post-ovulation period the corpus luteum disintegrates due to a lack of human chorionic gonadotropin (hCG). This would normally be produced by the embryo in the effort of maintaining progesterone and estrogen levels during pregnancy.
Therapeutically, clomifene is given early in the menstrual cycle. It is typically prescribed beginning on day 3 and continuing for five days. By that time, FSH levels are rising steadily, causing the development of a few follicles. Follicles, in turn, produce the estrogen, which circulates in serum. In the presence of clomifene, the body perceives a low level of estrogen, similar to day 22 in the previous cycle. Since estrogen can no longer effectively exert negative feedback on the hypothalamus, GnRH secretion becomes more rapidly pulsatile, which results in increased pituitary gonadotropin release. (More rapid, lower amplitude pulses of GnRH lead to increased LH and FSH secretion, while more irregular, larger amplitude pulses of GnRH leads to a decrease in the ratio of LH to FSH.) Increased FSH levels cause the growth of more ovarian follicles, and subsequently rupture of follicles resulting in ovulation. Ovulation occurs most often 6 to 7 days after a course of clomifene.
In normal men, 50 mg/day clomifene for 8 months has been found to increase testosterone levels by around 870 ng/dL in younger men and by around 490 ng/dL in elderly men.Estradiol levels increased by 62 pg/mL in younger men and by 40 pg/mL in elderly men. These findings suggest that the progonadotropic effects of clomifene are stronger in younger men than in older men. In men with hypogonadism, clomifene has been found to increase testosterone levels by 293 to 362 ng/dL and estradiol levels by 5.5 to 13 pg/mL. In a large clinical study of men with low testosterone levels (<400 ng/dL), 25 mg/day clomifene increased testosterone levels from 309 ng/dL to 642 ng/dL after 3 months of therapy. No significant changes in HDL cholesterol, triglycerides, fasting glucose, or prolactin levels were observed, although total cholesterol levels decreased significantly.
| Medication | Breast | Bone | Liver | Uterus | Vagina | Brain | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lipids | Coagulation | SHBG | IGF-1 | Hot flashes | Gonadotropins | |||||||||
| Estradiol | + | + | + | + | + | + | + | + | + | + | ||||
| "Ideal SERM" | – | + | + | ± | ± | ± | – | + | + | ± | ||||
| Bazedoxifene | – | + | + | + | + | ? | – | ± | – | ? | ||||
| Clomifene | – | + | + | ? | + | + | – | ? | – | ± | ||||
| Lasofoxifene | – | + | + | + | ? | ? | ± | ± | – | ? | ||||
| Ospemifene | – | + | + | + | + | + | ± | ± | – | ± | ||||
| Raloxifene | – | + | + | + | + | + | ± | – | – | ± | ||||
| Tamoxifen | – | + | + | + | + | + | + | – | – | ± | ||||
| Toremifene | – | + | + | + | + | + | + | – | – | ± | ||||
| Effect: + = Estrogenic / agonistic. ± = Mixed or neutral. – = Antiestrogenic / antagonistic. Note: SERMs generally increase gonadotropin levels in hypogonadal and eugonadal men as well as premenopausal women (antiestrogenic) but decrease gonadotropin levels in postmenopausal women (estrogenic). Sources: See template. | ||||||||||||||
Other activities
Clomifene is an inhibitor of the conversion of desmosterol into cholesterol by the enzyme 24-dehydrocholesterol reductase. Concerns about possible induction of desmosterolosis and associated symptoms such as cataracts and ichthyosis with extended exposure precluded the use of clomifene in the treatment of breast cancer. Continuous use of clomifene has been found to increase desmosterol levels by 10% and continuous high doses of clomifene (200 mg/day) have been reported to produce visual disturbances.
Pharmacokinetics
Clomifene produces N-desmethylclomifene, clomifenoxide (clomifene N-oxide), 4-hydroxyclomifene, and 4-hydroxy-N-desmethylclomifene as metabolites. Clomifene is a prodrug most importantly of 4-hydroxyclomifene and 4-hydroxy-N-desmethylclomifene, which are the most active of its metabolites. In one study, the peak levels after a single 50 mg dose of clomifene were 20.37 nmol/L for clomifene, 0.95 nmol/L for 4-hydroxyclomifene, and 1.15 nmol/L for 4-hydroxy-N-desmethylclomifene.
Clomifene has an onset of action of 5 to 10 days following course of treatment and an elimination half-life about 4 - 7days. In one study, after a single 50 mg dose of clomifene, the half-life of clomifene was 128 hours (5.3 days), of 4-hydroxyclomifene was 13 hours, and of 4-hydroxy-N-desmethylclomifene was 15 hours. Individuals with the CYP2D6*10 allele showed longer half-lives for 4-hydroxyclomifene and 4-hydroxy-N-desmethylclomifene. Primarily due to differences in CYP2D6 genetics, steady state concentrations and individual response to clomifene are highly variable.
Most clomifene metabolism occurs in the liver, where it undergoes enterohepatic recirculation. Clomifene and its metabolites are excreted primarily through feces (42%), and excretion can occur up to 6 weeks after discontinuation.
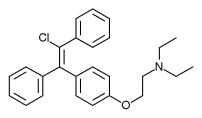
Chemistry
Clomifene is a triphenylethylene derivative. It is a mixture of two geometric isomers, the cis enclomifene ((E)-clomifene) form and trans zuclomifene ((Z)-clomifene) form. These two isomers contribute to the mixed estrogenic and antiestrogenic properties of clomifene. The typical ratio of these isomers after synthesis is 38% zuclomiphene and 62% enclomiphene. The United States Pharmacopeia specifies that clomifene preparations must contain between 30% and 50% zuclomiphene.
History
A team at William S. Merrell Chemical Company led by Frank Palopoli synthesized clomifene in 1956; after its biological activity was confirmed a patent was filed and issued in November 1959. Scientists at Merrell had previously synthesized chlorotrianisene and ethamoxytriphetol. Clomifene was studied in the treatment of advanced breast cancer during the period of 1964 to 1974 and was found to be effective but was abandoned due to concerns about desmosterolosis with extended use. Short-term use (e.g. days to months) did not raise the same concerns and clomifene continued to be studied for other indications.
| Antiestrogen | Dosage | Year(s) | Response rate | Toxicity |
|---|---|---|---|---|
| Ethamoxytriphetol | 500–4,500 mg/day | 1960 | 25% | Acute psychotic episodes |
| Clomifene | 100–300 mg/day | 1964–1974 | 34% | Fears of cataracts |
| Nafoxidine | 180–240 mg/day | 1976 | 31% | Cataracts, ichthyosis, photophobia |
| Tamoxifen | 20–40 mg/day | 1971–1973 | 31% | Transient thrombocytopeniaa |
| Footnotes: a = "The particular advantage of this drug is the low incidence of troublesome side effects (25)." "Side effects were usually trivial (26)." Sources: | ||||
Clinical studies were conducted under an Investigational New Drug Application; clomifene was third drug for which an IND had been filed under the 1962 Kefauver Harris Amendment to the Federal Food, Drug, and Cosmetic Act that had been passed in response to the thalidomide tragedy. It was approved for marketing in 1967 under the brand name Clomid. It was first used to treat cases of oligomenorrhea but was expanded to include treatment of anovulation when women undergoing treatment had higher than expected rates of pregnancy.
The drug is widely considered to have been a revolution in the treatment of female infertility, the beginning of the modern era of assisted reproductive technology, and the beginning of what in the words of Eli Y. Adashi, was "the onset of the US multiple births epidemic".
The company was acquired by Dow Chemical in 1980, and in 1989 Dow Chemical acquired 67 percent interest of Marion Laboratories, which was renamed Marion Merrell Dow. In 1995 Hoechst AG acquired the pharmaceutical business of Marion Merrell Dow. Hoechst in turn became part of Aventis in 1999, and subsequently a part of Sanofi. It became the most widely prescribed drug for ovulation induction to reverse anovulation or oligoovulation.
Society and culture
Brand names
Clomifene is marketed under many brand names worldwide, including Beclom, Bemot, Biogen, Blesifen, Chloramiphene, Clofert, Clomene, ClomHEXAL, Clomi, Clomid, Clomidac, Clomifen, Clomifencitrat, Clomifene, Clomifène, Clomifene citrate, Clomifeni citras, Clomifeno, Clomifert, Clomihexal, Clomiphen, Clomiphene, Clomiphene Citrate, Cloninn, Clostil, Clostilbegyt, Clovertil, Clovul, Dipthen, Dufine, Duinum, Fensipros, Fertab, Fertec, Fertex, Ferticlo, Fertil, Fertilan, Fertilphen, Fertin, Fertomid, Ferton, Fertotab, Fertyl, Fetrop, Folistim, Genoclom, Genozym, Hete, I-Clom, Ikaclomin, Klofit, Klomen, Klomifen, Lomifen, MER 41, Milophene, Ofertil, Omifin, Ova-mit, Ovamit, Ovinum, Ovipreg, Ovofar, Ovuclon, Ovulet, Pergotime, Pinfetil, Profertil, Prolifen, Provula, Reomen, Serofene, Serophene, Serpafar, Serpafar, Surole, Tocofeno, and Zimaquin.
Regulation
Clomifene is included on the World Anti-Doping Agency list of illegal doping agents in sport. It is listed because it is an "anti-estrogenic substance".
Research
Clomifene has been used almost exclusively for ovulation induction in premenopausal women, and has been studied very limitedly in postmenopausal women.
Clomifene was studied for treatment and prevention of breast cancer, but issues with toxicity led to abandonment of this indication, as did the discovery of tamoxifen. Like the structurally related drug triparanol, clomifene is known to inhibit the enzyme 24-dehydrocholesterol reductase and increase circulating desmosterol levels, making it unfavorable for extended use in breast cancer due to risk of side effects like irreversible cataracts.
External links
- "Clomifene". Drug Information Portal. U.S. National Library of Medicine.