
| Epothilones | ||||
|---|---|---|---|---|

Epothilones A (R = H) and B (R = CH3) | ||||
| Chemical formulae |
A: C26H39NO6S |
|||
| Molecular masses |
A: 493.66 g/mol |
|||
| CAS numbers |
A: 152044-53-6 |
|||
| PubChem |
A: 448799 |
|||
Epothilones C (R = H) and D (R = CH3) | ||||
| Chemical formulae |
C: C26H39NO5S |
|||
| Molecular masses |
C: 477.66 g/mol |
|||
| CAS numbers |
C: 186692-73-9 |
|||
| PubChem |
C: 9891226 |
|||

Epothilones E (R = H) and F (R = CH3) | ||||
| Chemical formulae |
E: C26H39NO7S |
|||
| Molecular masses |
E: 509.66 g/mol |
|||
| CAS numbers |
E: 201049-37-8 |
|||
| PubChem |
E: 9806341 |
Disclaimer and references | ||
Epothilones are a class of potential cancer drugs. Like taxanes, they prevent cancer cells from dividing by interfering with tubulin, but in early trials, epothilones have better efficacy and milder adverse effects than taxanes.
As of September 2008, epothilones A to F have been identified and characterized. Early studies in cancer cell lines and human cancer patients indicate superior efficacy to the taxanes. Their mechanism of action is similar, but their chemical structure is simpler. Due to their better water solubility, cremophors (solubilizing agents used for paclitaxel which can affect cardiac function and cause severe hypersensitivity) are not needed. Endotoxin-like properties known from paclitaxel, like activation of macrophages synthesizing inflammatory cytokines and nitric oxide, are not observed for epothilone B.
Epothilones were originally identified as metabolites produced by the soil-dwelling myxobacterium Sorangium cellulosum.
History
The structure of epothilone A was determined in 1996 using x-ray crystallography.
Mechanism of action
The principal mechanism of the epothilone class is the inhibition of the microtubule function. Microtubules are essential to cell division, and epothilones, therefore, stop cells from properly dividing. Epothilone B possesses the same biological effects as paclitaxel both in vitro and in cultured cells. This is because they share the same binding site, as well as binding affinity to the microtubule. Like paclitaxel, epothilone B binds to the αβ-tubulin heterodimer subunit. Once bound, the rate of αβ-tubulin dissociation decreases, thus stabilizing the microtubules. Furthermore, epothilone B has also been shown to induce tubulin polymerization into microtubules without the presence of GTP. This is caused by the formation of microtubule bundles throughout the cytoplasm. Finally, epothilone B also causes cell cycle arrest at the G2-M transition phase, thus leading to cytotoxicity and eventually cell apoptosis. The ability of epothilone to inhibit spindle function is generally attributed to its suppression of microtubule dynamics; but recent studies have demonstrated that suppression of dynamics occurs at concentrations lower than those needed to block mitosis. At higher antimitotic concentrations, paclitaxel appears to act by suppressing microtubule detachment from centrosomes, a process that is normally activated during mitosis. It is quite possible that epothilone can also act through a similar mechanism.
Analogs approved for medical use
One analog, ixabepilone, was approved in October 2007 by the United States Food and Drug Administration for use in the treatment of aggressive metastatic or locally advanced breast cancer that no longer responds to currently available chemotherapies. In November 2008, the EMEA refused a marketing authorization for Ixabepilone.
Clinical trials
Several synthetic epothilone analogs are currently undergoing clinical development for the treatment of various cancers.
Epothilone B has proven to contain potent in vivo anticancer activities at tolerated dose levels in several human xenograft models. As a result, epothilone B (patupilone) and various analogs are As of 2001 undergoing various clinical phases: patupilone and the fully synthetic sagopilone [SH-Y03757A, ZK-EPO, chemical structure] are in phase II trials; BMS-310705 in phase I trials).
Results of a phase III trial with ixabepilone (BMS-247550) in combination with capecitabine in metastatic breast cancer have been announced (2007 – leading to FDA approval).
Patupilone failed a phase III trial for ovarian cancer in 2010.
Utidelone is a genetically engineered epothilone analog that has shown benefits in a phase III breast cancer trial when added to capecitabine.
Total synthesis
Due to the high potency and clinical need for cancer treatments, epothilones have been the target of many total syntheses. The first group to publish the total synthesis of epothilones was S. J. Danishefsky et al. in 1996. This total synthesis of epothilone A was achieved via an intramolecular ester enolate-aldehyde condensation. Other syntheses of epothilones have been published by Nicolaou, Schinzer,Mulzer, and Carreira. In this approach, key building blocks aldehyde, glycidols, and ketoacid were constructed and coupled to the olefin metathesis precursor via an aldol reaction and then an esterification coupling. Grubbs' catalyst was employed to close the bis terminal olefin of the precursor compound. The resulting compounds were cis- and trans-macrocyclic isomers with distinct stereocenters. Epoxidation of cis- and trans-olefins yield epothilone A and its analogs.
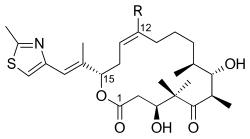
One of the total syntheses of epothilone B is outlined below and was described by the laboratory of K. C. Nicolaou. The retrosynthetic analysis revealed 1, 2, and 3 as the building blocks (Figure 1).
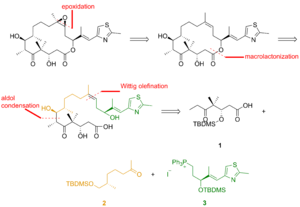
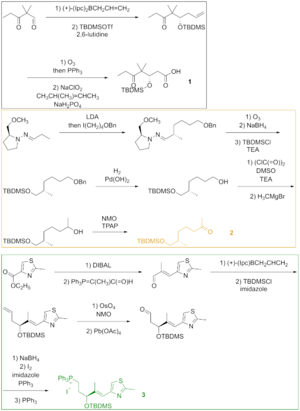
As seen in Figure 2, keto acid 1 was generated from the keto aldehyde that was converted to the silyl ether via asymmetric allylboration and silylation of the resulting alcohol. Ozonolysis of the silyl ether and Lindgren–Pinnick oxidation of the aldehyde afforded the keto acid. Ketone 2 was constructed via Enders alkylation starting from the hydrazone. Ozonolysis, the last step of the Enders alkylation, was followed by reduction of the aldehyde and silylation of the resulting alcohol. Hydrogenolysis of the benzyl ether gave the alcohol, which was oxidized under Swern condition and alkylated with the Grignard reagent to yield the secondary alcohol. Oxidation of this alcohol with the Ley–Griffith reagent gave the desired ketone. Thiazole 3 was synthesized from the ester, which was reduced with diisobutylaluminium hydride, and the aldehyde was reacted with the stabilized ylide in the Wittig reaction. Asymmetric allylboration of the α,β-unsaturated aldehyde and protection of the hydroxy group gave the silyl ether, whose terminal olefin was reacted with osmium tetroxide to a diol that was cleaved with lead tetraacetate to furnish the aldehyde. Reduction, iodination, and treatment with triphenylphosphine led to phosphonium salt.
Fragments 1, 2, and 3 were reacted with each other to deliver epothilone B in an approach including Wittig reaction, aldol reaction, and Yamaguchi esterification (Figure 3). Preparative thin-layer chromatography was used to separate the diastereomers.

Biosynthesis
Epothilone B is a 16-membered polyketide macrolactone with a methylthiazole group connected to the macrocycle by an olefinic bond. The polyketide backbone was synthesized by type I polyketide synthase (PKS) and the thiazole ring was derived from a cysteine incorporated by a nonribosomal peptide synthetase (NRPS). In this biosynthesis, both PKS and NRPS use carrier proteins, which have been post-translationally modified by phosphopantetheine groups, to join the growing chain. PKS uses coenzyme-A thioester to catalyze the reaction and modify the substrates by selectively reducing the β carbonyl to the hydroxyl (Ketoreductase, KR), the alkene (Dehydratase, DH), and the alkane (Enoyl Reductase, ER). PKS-I can also methylate the α carbon of the substrate. NRPS, on the other hand, uses amino acids activated on the enzyme as aminoacyl adenylates. Unlike PKS, epimerization, N-methylation, and heterocycle formation occurs in the NRPS enzyme.


Epothilone B starts with a 2-methyl-4-carboxythiazole starter unit, which was formed through the translational coupling between PKS, EPOS A (epoA) module, and NRPS, EPOS P(epoP) module. The EPOS A contains a modified β-ketoacyl-synthase (malonyl-ACP decarboxylase, KSQ), an acyltransferase (AT), an enoyl reductase (ER), and an acyl carrier protein domain (ACP). The EPOS P however, contains a heterocylization, an adenylation, an oxidase, and a thiolation domain. These domains are important because they are involved in the formation of the five-membered heterocyclic ring of thiazole. As seen in Figure 4, the EPOS P activates the cysteine and binds the activated cysteine as an aminoacyl-S-PCP. Once the cysteine has been bound, EPOS A loads an acetate unit onto the EPOS P complex, thus initiating the formation of the thiazoline ring by intramolecular cyclodehydration.
Once the 2-methylthiazole ring has been made, it is then transferred to the PKS EPOS B (epoB), EPOS C (epoC), EPOS D (epoD), EPOS E (epoE), and EPOS F (epoF) for subsequent elongation and modification to generate the olefinic bond, the 16-membered ring, and the epoxide, as seen in Figure 5. One important thing to note is the synthesis of the gem-dimethyl unit in module 7. These two dimethyls were not synthesized by two successive C-methylations. Instead, one of the methyl groups was derived from the propionate extender unit, while the second methyl group was integrated by a C-methyl-transferase domain.