

| Cell biology | |
|---|---|
| Animal cell diagram | |
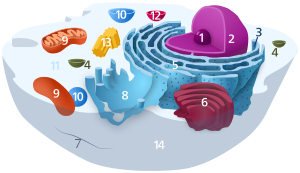 Components of a typical animal cell:
|
A mitochondrion (/ˌmaɪtəˈkɒndriən/;PL mitochondria) is an organelle found in the cells of most eukaryotes, such as animals, plants and fungi. Mitochondria have a double membrane structure and use aerobic respiration to generate adenosine triphosphate (ATP), which is used throughout the cell as a source of chemical energy. They were discovered by Albert von Kölliker in 1857 in the voluntary muscles of insects. The term mitochondrion was coined by Carl Benda in 1898. The mitochondrion is popularly nicknamed the "powerhouse of the cell", a phrase coined by Philip Siekevitz in a 1957 article of the same name.
Some cells in some multicellular organisms lack mitochondria (for example, mature mammalian red blood cells). A large number of unicellular organisms, such as microsporidia, parabasalids and diplomonads, have reduced or transformed their mitochondria into other structures. One eukaryote, Monocercomonoides, is known to have completely lost its mitochondria, and one multicellular organism, Henneguya salminicola, is known to have retained mitochondrion-related organelles in association with a complete loss of their mitochondrial genome.
Mitochondria are commonly between 0.75 and 3 μm2 in cross section, but vary considerably in size and structure. Unless specifically stained, they are not visible. In addition to supplying cellular energy, mitochondria are involved in other tasks, such as signaling, cellular differentiation, and cell death, as well as maintaining control of the cell cycle and cell growth.Mitochondrial biogenesis is in turn temporally coordinated with these cellular processes. Mitochondria have been implicated in several human disorders and conditions, such as mitochondrial diseases,cardiac dysfunction, heart failure and autism.
The number of mitochondria in a cell can vary widely by organism, tissue, and cell type. A mature red blood cell has no mitochondria, whereas a liver cell can have more than 2000. The mitochondrion is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, intermembrane space, inner membrane, cristae, and matrix.
Although most of a eukaryotic cell's DNA is contained in the cell nucleus, the mitochondrion has its own genome ("mitogenome") that is substantially similar to bacterial genomes. This finding has led to general acceptance of the endosymbiotic hypothesis - that free-living prokaryotic ancestors of modern mitochondria permanently fused with eukaryotic cells in the distant past, evolving such that modern animals, plants, fungi, and other eukaryotes are able to respire to generate cellular energy.
Structure

Mitochondria may have a number of different shapes. A mitochondrion contains outer and inner membranes composed of phospholipid bilayers and proteins. The two membranes have different properties. Because of this double-membraned organization, there are five distinct parts to a mitochondrion:
- The outer mitochondrial membrane,
- The intermembrane space (the space between the outer and inner membranes),
- The inner mitochondrial membrane,
- The cristae space (formed by infoldings of the inner membrane), and
- The matrix (space within the inner membrane), which is a fluid.
Mitochondria have folding to increase surface area, which in turn increases ATP (adenosine triphosphate) production. Mitochondria stripped of their outer membrane are called mitoplasts.
Outer membrane
The outer mitochondrial membrane, which encloses the entire organelle, is 60 to 75 angstroms (Å) thick. It has a protein-to-phospholipid ratio similar to that of the cell membrane (about 1:1 by weight). It contains large numbers of integral membrane proteins called porins. A major trafficking protein is the pore-forming voltage-dependent anion channel (VDAC). The VDAC is the primary transporter of nucleotides, ions and metabolites between the cytosol and the intermembrane space. It is formed as a beta barrel that spans the outer membrane, similar to that in the gram-negative bacterial membrane. Larger proteins can enter the mitochondrion if a signaling sequence at their N-terminus binds to a large multisubunit protein called translocase in the outer membrane, which then actively moves them across the membrane. Mitochondrial pro-proteins are imported through specialised translocation complexes.
The outer membrane also contains enzymes involved in such diverse activities as the elongation of fatty acids, oxidation of epinephrine, and the degradation of tryptophan. These enzymes include monoamine oxidase, rotenone-insensitive NADH-cytochrome c-reductase, kynurenine hydroxylase and fatty acid Co-A ligase. Disruption of the outer membrane permits proteins in the intermembrane space to leak into the cytosol, leading to cell death. The outer mitochondrial membrane can associate with the endoplasmic reticulum (ER) membrane, in a structure called MAM (mitochondria-associated ER-membrane). This is important in the ER-mitochondria calcium signaling and is involved in the transfer of lipids between the ER and mitochondria. Outside the outer membrane are small (diameter: 60 Å) particles named sub-units of Parson.
Intermembrane space
The mitochondrial intermembrane space is the space between the outer membrane and the inner membrane. It is also known as perimitochondrial space. Because the outer membrane is freely permeable to small molecules, the concentrations of small molecules, such as ions and sugars, in the intermembrane space is the same as in the cytosol. However, large proteins must have a specific signaling sequence to be transported across the outer membrane, so the protein composition of this space is different from the protein composition of the cytosol. One protein that is localized to the intermembrane space in this way is cytochrome c.
Inner membrane
The inner mitochondrial membrane contains proteins with three types of functions:
- Those that perform the electron transport chain redox reactions
- ATP synthase, which generates ATP in the matrix
- Specific transport proteins that regulate metabolite passage into and out of the mitochondrial matrix
It contains more than 151 different polypeptides, and has a very high protein-to-phospholipid ratio (more than 3:1 by weight, which is about 1 protein for 15 phospholipids). The inner membrane is home to around 1/5 of the total protein in a mitochondrion. Additionally, the inner membrane is rich in an unusual phospholipid, cardiolipin. This phospholipid was originally discovered in cow hearts in 1942, and is usually characteristic of mitochondrial and bacterial plasma membranes. Cardiolipin contains four fatty acids rather than two, and may help to make the inner membrane impermeable, and its disruption can lead to multiple clinical disorders including neurological disorders and cancer. Unlike the outer membrane, the inner membrane does not contain porins, and is highly impermeable to all molecules. Almost all ions and molecules require special membrane transporters to enter or exit the matrix. Proteins are ferried into the matrix via the translocase of the inner membrane (TIM) complex or via OXA1L. In addition, there is a membrane potential across the inner membrane, formed by the action of the enzymes of the electron transport chain. Inner membrane fusion is mediated by the inner membrane protein OPA1.
Cristae

The inner mitochondrial membrane is compartmentalized into numerous folds called cristae, which expand the surface area of the inner mitochondrial membrane, enhancing its ability to produce ATP. For typical liver mitochondria, the area of the inner membrane is about five times as large as that of the outer membrane. This ratio is variable and mitochondria from cells that have a greater demand for ATP, such as muscle cells, contain even more cristae. Mitochondria within the same cell can have substantially different crista-density, with the ones that are required to produce more energy having much more crista-membrane surface. These folds are studded with small round bodies known as F1 particles or oxysomes.
Matrix
The matrix is the space enclosed by the inner membrane. It contains about 2/3 of the total proteins in a mitochondrion. The matrix is important in the production of ATP with the aid of the ATP synthase contained in the inner membrane. The matrix contains a highly concentrated mixture of hundreds of enzymes, special mitochondrial ribosomes, tRNA, and several copies of the mitochondrial DNA genome. Of the enzymes, the major functions include oxidation of pyruvate and fatty acids, and the citric acid cycle. The DNA molecules are packaged into nucleoids by proteins, one of which is TFAM.
Function
The most prominent roles of mitochondria are to produce the energy currency of the cell, ATP (i.e., phosphorylation of ADP), through respiration and to regulate cellular metabolism. The central set of reactions involved in ATP production are collectively known as the citric acid cycle, or the Krebs cycle, and oxidative phosphorylation. However, the mitochondrion has many other functions in addition to the production of ATP.
Energy conversion
A dominant role for the mitochondria is the production of ATP, as reflected by the large number of proteins in the inner membrane for this task. This is done by oxidizing the major products of glucose: pyruvate, and NADH, which are produced in the cytosol. This type of cellular respiration, known as aerobic respiration, is dependent on the presence of oxygen. When oxygen is limited, the glycolytic products will be metabolized by anaerobic fermentation, a process that is independent of the mitochondria. The production of ATP from glucose and oxygen has an approximately 13-times higher yield during aerobic respiration compared to fermentation. Plant mitochondria can also produce a limited amount of ATP either by breaking the sugar produced during photosynthesis or without oxygen by using the alternate substrate nitrite. ATP crosses out through the inner membrane with the help of a specific protein, and across the outer membrane via porins. After conversion of ATP to ADP by dephosphorylation that releases energy, ADP returns via the same route.
Pyruvate and the citric acid cycle
Pyruvate molecules produced by glycolysis are actively transported across the inner mitochondrial membrane, and into the matrix where they can either be oxidized and combined with coenzyme A to form CO2, acetyl-CoA, and NADH, or they can be carboxylated (by pyruvate carboxylase) to form oxaloacetate. This latter reaction "fills up" the amount of oxaloacetate in the citric acid cycle and is therefore an anaplerotic reaction, increasing the cycle's capacity to metabolize acetyl-CoA when the tissue's energy needs (e.g., in muscle) are suddenly increased by activity.
In the citric acid cycle, all the intermediates (e.g. citrate, iso-citrate, alpha-ketoglutarate, succinate, fumarate, malate and oxaloacetate) are regenerated during each turn of the cycle. Adding more of any of these intermediates to the mitochondrion therefore means that the additional amount is retained within the cycle, increasing all the other intermediates as one is converted into the other. Hence, the addition of any one of them to the cycle has an anaplerotic effect, and its removal has a cataplerotic effect. These anaplerotic and cataplerotic reactions will, during the course of the cycle, increase or decrease the amount of oxaloacetate available to combine with acetyl-CoA to form citric acid. This in turn increases or decreases the rate of ATP production by the mitochondrion, and thus the availability of ATP to the cell.
Acetyl-CoA, on the other hand, derived from pyruvate oxidation, or from the beta-oxidation of fatty acids, is the only fuel to enter the citric acid cycle. With each turn of the cycle one molecule of acetyl-CoA is consumed for every molecule of oxaloacetate present in the mitochondrial matrix, and is never regenerated. It is the oxidation of the acetate portion of acetyl-CoA that produces CO2 and water, with the energy thus released captured in the form of ATP.
In the liver, the carboxylation of cytosolic pyruvate into intra-mitochondrial oxaloacetate is an early step in the gluconeogenic pathway, which converts lactate and de-aminated alanine into glucose, under the influence of high levels of glucagon and/or epinephrine in the blood. Here, the addition of oxaloacetate to the mitochondrion does not have a net anaplerotic effect, as another citric acid cycle intermediate (malate) is immediately removed from the mitochondrion to be converted to cytosolic oxaloacetate, and ultimately to glucose, in a process that is almost the reverse of glycolysis.
The enzymes of the citric acid cycle are located in the mitochondrial matrix, with the exception of succinate dehydrogenase, which is bound to the inner mitochondrial membrane as part of Complex II. The citric acid cycle oxidizes the acetyl-CoA to carbon dioxide, and, in the process, produces reduced cofactors (three molecules of NADH and one molecule of FADH2) that are a source of electrons for the electron transport chain, and a molecule of GTP (which is readily converted to an ATP).
O2 and NADH: energy-releasing reactions
The electrons from NADH and FADH2 are transferred to oxygen (O2) and hydrogen (protons) in several steps via an electron transport chain. NADH and FADH2 molecules are produced within the matrix via the citric acid cycle and in the cytoplasm by glycolysis. Reducing equivalents from the cytoplasm can be imported via the malate-aspartate shuttle system of antiporter proteins or fed into the electron transport chain using a glycerol phosphate shuttle.
The major energy-releasing reactions that make the mitochondrion the "powerhouse of the cell" occur at protein complexes I, III and IV in the inner mitochondrial membrane (NADH dehydrogenase (ubiquinone), cytochrome c reductase, and cytochrome c oxidase). At complex IV, O2 reacts with the reduced form of iron in cytochrome c:


releasing a lot of free energy from the reactants without breaking bonds of an organic fuel. The free energy put in to remove an electron from Fe2+ is released at complex III when Fe3+ of cytochrome c reacts to oxidize ubiquinol (QH2):


The ubiquinone (Q) generated reacts, in complex I, with NADH:


While the reactions are controlled by an electron transport chain, free electrons are not amongst the reactants or products in the three reactions shown and therefore do not affect the free energy released, which is used to pump protons (H+) into the intermembrane space. This process is efficient, but a small percentage of electrons may prematurely reduce oxygen, forming reactive oxygen species such as superoxide. This can cause oxidative stress in the mitochondria and may contribute to the decline in mitochondrial function associated with aging.
As the proton concentration increases in the intermembrane space, a strong electrochemical gradient is established across the inner membrane. The protons can return to the matrix through the ATP synthase complex, and their potential energy is used to synthesize ATP from ADP and inorganic phosphate (Pi). This process is called chemiosmosis, and was first described by Peter Mitchell, who was awarded the 1978 Nobel Prize in Chemistry for his work. Later, part of the 1997 Nobel Prize in Chemistry was awarded to Paul D. Boyer and John E. Walker for their clarification of the working mechanism of ATP synthase.
Heat production
Under certain conditions, protons can re-enter the mitochondrial matrix without contributing to ATP synthesis. This process is known as proton leak or mitochondrial uncoupling and is due to the facilitated diffusion of protons into the matrix. The process results in the unharnessed potential energy of the proton electrochemical gradient being released as heat. The process is mediated by a proton channel called thermogenin, or UCP1. Thermogenin is primarily found in brown adipose tissue, or brown fat, and is responsible for non-shivering thermogenesis. Brown adipose tissue is found in mammals, and is at its highest levels in early life and in hibernating animals. In humans, brown adipose tissue is present at birth and decreases with age.
Storage of calcium ions

The concentrations of free calcium in the cell can regulate an array of reactions and is important for signal transduction in the cell. Mitochondria can transiently store calcium, a contributing process for the cell's homeostasis of calcium. Their ability to rapidly take in calcium for later release makes them good "cytosolic buffers" for calcium. The endoplasmic reticulum (ER) is the most significant storage site of calcium, and there is a significant interplay between the mitochondrion and ER with regard to calcium. The calcium is taken up into the matrix by the mitochondrial calcium uniporter on the inner mitochondrial membrane. It is primarily driven by the mitochondrial membrane potential. Release of this calcium back into the cell's interior can occur via a sodium-calcium exchange protein or via "calcium-induced-calcium-release" pathways. This can initiate calcium spikes or calcium waves with large changes in the membrane potential. These can activate a series of second messenger system proteins that can coordinate processes such as neurotransmitter release in nerve cells and release of hormones in endocrine cells.
Ca2+ influx to the mitochondrial matrix has recently been implicated as a mechanism to regulate respiratory bioenergetics by allowing the electrochemical potential across the membrane to transiently "pulse" from ΔΨ-dominated to pH-dominated, facilitating a reduction of oxidative stress. In neurons, concomitant increases in cytosolic and mitochondrial calcium act to synchronize neuronal activity with mitochondrial energy metabolism. Mitochondrial matrix calcium levels can reach the tens of micromolar levels, which is necessary for the activation of isocitrate dehydrogenase, one of the key regulatory enzymes of the Krebs cycle.
Cellular proliferation regulation
The relationship between cellular proliferation and mitochondria has been investigated. Tumor cells require ample ATP to synthesize bioactive compounds such as lipids, proteins, and nucleotides for rapid proliferation. The majority of ATP in tumor cells is generated via the oxidative phosphorylation pathway (OxPhos). Interference with OxPhos cause cell cycle arrest suggesting that mitochondria play a role in cell proliferation. Mitochondrial ATP production is also vital for cell division and differentiation in infection in addition to basic functions in the cell including the regulation of cell volume, solute concentration, and cellular architecture. ATP levels differ at various stages of the cell cycle suggesting that there is a relationship between the abundance of ATP and the cell's ability to enter a new cell cycle. ATP's role in the basic functions of the cell make the cell cycle sensitive to changes in the availability of mitochondrial derived ATP. The variation in ATP levels at different stages of the cell cycle support the hypothesis that mitochondria play an important role in cell cycle regulation. Although the specific mechanisms between mitochondria and the cell cycle regulation is not well understood, studies have shown that low energy cell cycle checkpoints monitor the energy capability before committing to another round of cell division.
Additional functions
Mitochondria play a central role in many other metabolic tasks, such as:
- Signaling through mitochondrial reactive oxygen species
- Regulation of the membrane potential
- Apoptosis-programmed cell death
- Calcium signaling (including calcium-evoked apoptosis)
- Regulation of cellular metabolism
- Certain heme synthesis reactions (see also: Porphyrin)
- Steroid synthesis
- Hormonal signaling – mitochondria are sensitive and responsive to hormones, in part by the action of mitochondrial estrogen receptors (mtERs). These receptors have been found in various tissues and cell types, including brain and heart
- Immune signaling
- Neuronal mitochondria also contribute to cellular quality control by reporting neuronal status towards microglia through specialised somatic-junctions.
- Mitochondria of developing neurons contribute to intercellular signaling towards microglia, which communication is indispensable for proper regulation of brain development.
Some mitochondrial functions are performed only in specific types of cells. For example, mitochondria in liver cells contain enzymes that allow them to detoxify ammonia, a waste product of protein metabolism. A mutation in the genes regulating any of these functions can result in mitochondrial diseases.
Mitochondrial proteins (proteins transcribed from mitochondrial DNA) vary depending on the tissue and the species. In humans, 615 distinct types of proteins have been identified from cardiac mitochondria, whereas in rats, 940 proteins have been reported. The mitochondrial proteome is thought to be dynamically regulated.
Organization and distribution

Mitochondria (or related structures) are found in all eukaryotes (except the Oxymonad Monocercomonoides). Although commonly depicted as bean-like structures they form a highly dynamic network in the majority of cells where they constantly undergo fission and fusion. The population of all the mitochondria of a given cell constitutes the chondriome. Mitochondria vary in number and location according to cell type. A single mitochondrion is often found in unicellular organisms, while human liver cells have about 1000–2000 mitochondria per cell, making up 1/5 of the cell volume. The mitochondrial content of otherwise similar cells can vary substantially in size and membrane potential, with differences arising from sources including uneven partitioning at cell division, leading to extrinsic differences in ATP levels and downstream cellular processes. The mitochondria can be found nestled between myofibrils of muscle or wrapped around the sperm flagellum. Often, they form a complex 3D branching network inside the cell with the cytoskeleton. The association with the cytoskeleton determines mitochondrial shape, which can affect the function as well: different structures of the mitochondrial network may afford the population a variety of physical, chemical, and signalling advantages or disadvantages. Mitochondria in cells are always distributed along microtubules and the distribution of these organelles is also correlated with the endoplasmic reticulum. Recent evidence suggests that vimentin, one of the components of the cytoskeleton, is also critical to the association with the cytoskeleton.
Mitochondria-associated ER membrane (MAM)
The mitochondria-associated ER membrane (MAM) is another structural element that is increasingly recognized for its critical role in cellular physiology and homeostasis. Once considered a technical snag in cell fractionation techniques, the alleged ER vesicle contaminants that invariably appeared in the mitochondrial fraction have been re-identified as membranous structures derived from the MAM—the interface between mitochondria and the ER. Physical coupling between these two organelles had previously been observed in electron micrographs and has more recently been probed with fluorescence microscopy. Such studies estimate that at the MAM, which may comprise up to 20% of the mitochondrial outer membrane, the ER and mitochondria are separated by a mere 10–25 nm and held together by protein tethering complexes.
Purified MAM from subcellular fractionation is enriched in enzymes involved in phospholipid exchange, in addition to channels associated with Ca2+ signaling. These hints of a prominent role for the MAM in the regulation of cellular lipid stores and signal transduction have been borne out, with significant implications for mitochondrial-associated cellular phenomena, as discussed below. Not only has the MAM provided insight into the mechanistic basis underlying such physiological processes as intrinsic apoptosis and the propagation of calcium signaling, but it also favors a more refined view of the mitochondria. Though often seen as static, isolated 'powerhouses' hijacked for cellular metabolism through an ancient endosymbiotic event, the evolution of the MAM underscores the extent to which mitochondria have been integrated into overall cellular physiology, with intimate physical and functional coupling to the endomembrane system.
Phospholipid transfer
The MAM is enriched in enzymes involved in lipid biosynthesis, such as phosphatidylserine synthase on the ER face and phosphatidylserine decarboxylase on the mitochondrial face. Because mitochondria are dynamic organelles constantly undergoing fission and fusion events, they require a constant and well-regulated supply of phospholipids for membrane integrity. But mitochondria are not only a destination for the phospholipids they finish synthesis of; rather, this organelle also plays a role in inter-organelle trafficking of the intermediates and products of phospholipid biosynthetic pathways, ceramide and cholesterol metabolism, and glycosphingolipid anabolism.
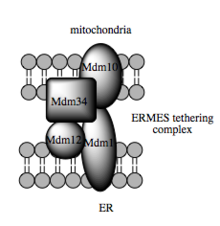
Such trafficking capacity depends on the MAM, which has been shown to facilitate transfer of lipid intermediates between organelles. In contrast to the standard vesicular mechanism of lipid transfer, evidence indicates that the physical proximity of the ER and mitochondrial membranes at the MAM allows for lipid flipping between opposed bilayers. Despite this unusual and seemingly energetically unfavorable mechanism, such transport does not require ATP. Instead, in yeast, it has been shown to be dependent on a multiprotein tethering structure termed the ER-mitochondria encounter structure, or ERMES, although it remains unclear whether this structure directly mediates lipid transfer or is required to keep the membranes in sufficiently close proximity to lower the energy barrier for lipid flipping.
The MAM may also be part of the secretory pathway, in addition to its role in intracellular lipid trafficking. In particular, the MAM appears to be an intermediate destination between the rough ER and the Golgi in the pathway that leads to very-low-density lipoprotein, or VLDL, assembly and secretion. The MAM thus serves as a critical metabolic and trafficking hub in lipid metabolism.
Calcium signaling
A critical role for the ER in calcium signaling was acknowledged before such a role for the mitochondria was widely accepted, in part because the low affinity of Ca2+ channels localized to the outer mitochondrial membrane seemed to contradict this organelle's purported responsiveness to changes in intracellular Ca2+ flux. But the presence of the MAM resolves this apparent contradiction: the close physical association between the two organelles results in Ca2+ microdomains at contact points that facilitate efficient Ca2+ transmission from the ER to the mitochondria. Transmission occurs in response to so-called "Ca2+ puffs" generated by spontaneous clustering and activation of IP3R, a canonical ER membrane Ca2+ channel.
The fate of these puffs—in particular, whether they remain restricted to isolated locales or integrated into Ca2+ waves for propagation throughout the cell—is determined in large part by MAM dynamics. Although reuptake of Ca2+ by the ER (concomitant with its release) modulates the intensity of the puffs, thus insulating mitochondria to a certain degree from high Ca2+ exposure, the MAM often serves as a firewall that essentially buffers Ca2+ puffs by acting as a sink into which free ions released into the cytosol can be funneled. This Ca2+ tunneling occurs through the low-affinity Ca2+ receptor VDAC1, which recently has been shown to be physically tethered to the IP3R clusters on the ER membrane and enriched at the MAM. The ability of mitochondria to serve as a Ca2+ sink is a result of the electrochemical gradient generated during oxidative phosphorylation, which makes tunneling of the cation an exergonic process. Normal, mild calcium influx from cytosol into the mitochondrial matrix causes transient depolarization that is corrected by pumping out protons.
But transmission of Ca2+ is not unidirectional; rather, it is a two-way street. The properties of the Ca2+ pump SERCA and the channel IP3R present on the ER membrane facilitate feedback regulation coordinated by MAM function. In particular, the clearance of Ca2+ by the MAM allows for spatio-temporal patterning of Ca2+ signaling because Ca2+ alters IP3R activity in a biphasic manner.SERCA is likewise affected by mitochondrial feedback: uptake of Ca2+ by the MAM stimulates ATP production, thus providing energy that enables SERCA to reload the ER with Ca2+ for continued Ca2+ efflux at the MAM. Thus, the MAM is not a passive buffer for Ca2+ puffs; rather it helps modulate further Ca2+ signaling through feedback loops that affect ER dynamics.
Regulating ER release of Ca2+ at the MAM is especially critical because only a certain window of Ca2+ uptake sustains the mitochondria, and consequently the cell, at homeostasis. Sufficient intraorganelle Ca2+ signaling is required to stimulate metabolism by activating dehydrogenase enzymes critical to flux through the citric acid cycle. However, once Ca2+ signaling in the mitochondria passes a certain threshold, it stimulates the intrinsic pathway of apoptosis in part by collapsing the mitochondrial membrane potential required for metabolism. Studies examining the role of pro- and anti-apoptotic factors support this model; for example, the anti-apoptotic factor Bcl-2 has been shown to interact with IP3Rs to reduce Ca2+ filling of the ER, leading to reduced efflux at the MAM and preventing collapse of the mitochondrial membrane potential post-apoptotic stimuli. Given the need for such fine regulation of Ca2+ signaling, it is perhaps unsurprising that dysregulated mitochondrial Ca2+ has been implicated in several neurodegenerative diseases, while the catalogue of tumor suppressors includes a few that are enriched at the MAM.
Molecular basis for tethering
Recent advances in the identification of the tethers between the mitochondrial and ER membranes suggest that the scaffolding function of the molecular elements involved is secondary to other, non-structural functions. In yeast, ERMES, a multiprotein complex of interacting ER- and mitochondrial-resident membrane proteins, is required for lipid transfer at the MAM and exemplifies this principle. One of its components, for example, is also a constituent of the protein complex required for insertion of transmembrane beta-barrel proteins into the lipid bilayer. However, a homologue of the ERMES complex has not yet been identified in mammalian cells. Other proteins implicated in scaffolding likewise have functions independent of structural tethering at the MAM; for example, ER-resident and mitochondrial-resident mitofusins form heterocomplexes that regulate the number of inter-organelle contact sites, although mitofusins were first identified for their role in fission and fusion events between individual mitochondria.Glucose-related protein 75 (grp75) is another dual-function protein. In addition to the matrix pool of grp75, a portion serves as a chaperone that physically links the mitochondrial and ER Ca2+ channels VDAC and IP3R for efficient Ca2+ transmission at the MAM. Another potential tether is Sigma-1R, a non-opioid receptor whose stabilization of ER-resident IP3R may preserve communication at the MAM during the metabolic stress response.
Perspective
The MAM is a critical signaling, metabolic, and trafficking hub in the cell that allows for the integration of ER and mitochondrial physiology. Coupling between these organelles is not simply structural but functional as well and critical for overall cellular physiology and homeostasis. The MAM thus offers a perspective on mitochondria that diverges from the traditional view of this organelle as a static, isolated unit appropriated for its metabolic capacity by the cell. Instead, this mitochondrial-ER interface emphasizes the integration of the mitochondria, the product of an endosymbiotic event, into diverse cellular processes. Recently it has also been shown, that mitochondria and MAM-s in neurons are anchored to specialised intercellular communication sites (so called somatic-junctions). Microglial processes monitor and protect neuronal functions at these sites, and MAM-s are supposed to have an important role in this type of cellular quality-control.
Origin and evolution
There are two hypotheses about the origin of mitochondria: endosymbiotic and autogenous. The endosymbiotic hypothesis suggests that mitochondria were originally prokaryotic cells, capable of implementing oxidative mechanisms that were not possible for eukaryotic cells; they became endosymbionts living inside the eukaryote. In the autogenous hypothesis, mitochondria were born by splitting off a portion of DNA from the nucleus of the eukaryotic cell at the time of divergence with the prokaryotes; this DNA portion would have been enclosed by membranes, which could not be crossed by proteins. Since mitochondria have many features in common with bacteria, the endosymbiotic hypothesis is the more widely accepted of the two accounts.
A mitochondrion contains DNA, which is organized as several copies of a single, usually circular chromosome. This mitochondrial chromosome contains genes for redox proteins, such as those of the respiratory chain. The CoRR hypothesis proposes that this co-location is required for redox regulation. The mitochondrial genome codes for some RNAs of ribosomes, and the 22 tRNAs necessary for the translation of mRNAs into protein. The circular structure is also found in prokaryotes. The proto-mitochondrion was probably closely related to Rickettsia. However, the exact relationship of the ancestor of mitochondria to the alphaproteobacteria and whether the mitochondrion was formed at the same time or after the nucleus, remains controversial. For example, it has been suggested that the SAR11 clade of bacteria shares a relatively recent common ancestor with the mitochondria, while phylogenomic analyses indicate that mitochondria evolved from a Pseudomonadota lineage that is closely related to or a member of alphaproteobacteria. Some papers describe mitochondria as sister to the alphaproteobactera, together forming the sister the marineproteo1 group, together forming the sister to Magnetococcidae.
| Proteobacteria |
|
||||||||||||||||||||||||
The ribosomes coded for by the mitochondrial DNA are similar to those from bacteria in size and structure. They closely resemble the bacterial 70S ribosome and not the 80S cytoplasmic ribosomes, which are coded for by nuclear DNA.
The endosymbiotic relationship of mitochondria with their host cells was popularized by Lynn Margulis. The endosymbiotic hypothesis suggests that mitochondria descended from aerobic bacteria that somehow survived endocytosis by another cell, and became incorporated into the cytoplasm. The ability of these bacteria to conduct respiration in host cells that had relied on glycolysis and fermentation would have provided a considerable evolutionary advantage. This symbiotic relationship probably developed 1.7 to 2 billion years ago.

A few groups of unicellular eukaryotes have only vestigial mitochondria or derived structures: The microsporidians, metamonads, and archamoebae. These groups appear as the most primitive eukaryotes on phylogenetic trees constructed using rRNA information, which once suggested that they appeared before the origin of mitochondria. However, this is now known to be an artifact of long-branch attraction: They are derived groups and retain genes or organelles derived from mitochondria (e. g., mitosomes and hydrogenosomes). Hydrogenosomes, mitosomes, and related organelles as found in some loricifera (e. g. Spinoloricus) and myxozoa (e. g. Henneguya zschokkei) are together classified as MROs, mitochondrion-related organelles.
Monocercomonoides appear to have lost their mitochondria completely and at least some of the mitochondrial functions seem to be carried out by cytoplasmic proteins now.
Mitochondrial genetics

Mitochondria contain their own genome. The human mitochondrial genome is a circular double-stranded DNA molecule of about 16 kilobases. It encodes 37 genes: 13 for subunits of respiratory complexes I, III, IV and V, 22 for mitochondrial tRNA (for the 20 standard amino acids, plus an extra gene for leucine and serine), and 2 for rRNA (12S and 16S rRNA). One mitochondrion can contain two to ten copies of its DNA. One of the two mitochondrial DNA (mtDNA) strands has a disproportionately higher ratio of the heavier nucleotides adenine and guanine, and this is termed the heavy strand (or H strand), whereas the other strand is termed the light strand (or L strand). The weight difference allows the two strands to be separated by centrifugation. mtDNA has one long non-coding stretch known as the non-coding region (NCR), which contains the heavy strand promoter (HSP) and light strand promoter (LSP) for RNA transcription, the origin of replication for the H strand (OriH) localized on the L strand, three conserved sequence boxes (CSBs 1–3), and a termination-associated sequence (TAS). The origin of replication for the L strand (OriL) is localized on the H strand 11,000 bp downstream of OriH, located within a cluster of genes coding for tRNA.
As in prokaryotes, there is a very high proportion of coding DNA and an absence of repeats. Mitochondrial genes are transcribed as multigenic transcripts, which are cleaved and polyadenylated to yield mature mRNAs. Most proteins necessary for mitochondrial function are encoded by genes in the cell nucleus and the corresponding proteins are imported into the mitochondrion. The exact number of genes encoded by the nucleus and the mitochondrial genome differs between species. Most mitochondrial genomes are circular. In general, mitochondrial DNA lacks introns, as is the case in the human mitochondrial genome; however, introns have been observed in some eukaryotic mitochondrial DNA, such as that of yeast and protists, including Dictyostelium discoideum. Between protein-coding regions, tRNAs are present. Mitochondrial tRNA genes have different sequences from the nuclear tRNAs, but lookalikes of mitochondrial tRNAs have been found in the nuclear chromosomes with high sequence similarity.
In animals, the mitochondrial genome is typically a single circular chromosome that is approximately 16 kb long and has 37 genes. The genes, while highly conserved, may vary in location. Curiously, this pattern is not found in the human body louse (Pediculus humanus). Instead, this mitochondrial genome is arranged in 18 minicircular chromosomes, each of which is 3–4 kb long and has one to three genes. This pattern is also found in other sucking lice, but not in chewing lice. Recombination has been shown to occur between the minichromosomes.
Human population genetic studies
The near-absence of genetic recombination in mitochondrial DNA makes it a useful source of information for studying population genetics and evolutionary biology. Because all the mitochondrial DNA is inherited as a single unit, or haplotype, the relationships between mitochondrial DNA from different individuals can be represented as a gene tree. Patterns in these gene trees can be used to infer the evolutionary history of populations. The classic example of this is in human evolutionary genetics, where the molecular clock can be used to provide a recent date for mitochondrial Eve. This is often interpreted as strong support for a recent modern human expansion out of Africa. Another human example is the sequencing of mitochondrial DNA from Neanderthal bones. The relatively large evolutionary distance between the mitochondrial DNA sequences of Neanderthals and living humans has been interpreted as evidence for the lack of interbreeding between Neanderthals and modern humans.
However, mitochondrial DNA reflects only the history of the females in a population. This can be partially overcome by the use of paternal genetic sequences, such as the non-recombining region of the Y-chromosome.
Recent measurements of the molecular clock for mitochondrial DNA reported a value of 1 mutation every 7884 years dating back to the most recent common ancestor of humans and apes, which is consistent with estimates of mutation rates of autosomal DNA (10−8 per base per generation).
Alternative genetic code
| Organism | Codon | Standard | Mitochondria |
|---|---|---|---|
| Mammals | AGA, AGG | Arginine | Stop codon |
| Invertebrates | AGA, AGG | Arginine | Serine |
| Fungi | CUA | Leucine | Threonine |
| All of the above | AUA | Isoleucine | Methionine |
| UGA | Stop codon | Tryptophan |
While slight variations on the standard genetic code had been predicted earlier, none was discovered until 1979, when researchers studying human mitochondrial genes determined that they used an alternative code. Nonetheless, the mitochondria of many other eukaryotes, including most plants, use the standard code. Many slight variants have been discovered since, including various alternative mitochondrial codes. Further, the AUA, AUC, and AUU codons are all allowable start codons.
Some of these differences should be regarded as pseudo-changes in the genetic code due to the phenomenon of RNA editing, which is common in mitochondria. In higher plants, it was thought that CGG encoded for tryptophan and not arginine; however, the codon in the processed RNA was discovered to be the UGG codon, consistent with the standard genetic code for tryptophan. Of note, the arthropod mitochondrial genetic code has undergone parallel evolution within a phylum, with some organisms uniquely translating AGG to lysine.
Replication and inheritance
Mitochondria divide by mitochondrial fission, a form of binary fission that is also done by bacteria although the process is tightly regulated by the host eukaryotic cell and involves communication between and contact with several other organelles. The regulation of this division differs between eukaryotes. In many single-celled eukaryotes, their growth and division are linked to the cell cycle. For example, a single mitochondrion may divide synchronously with the nucleus. This division and segregation process must be tightly controlled so that each daughter cell receives at least one mitochondrion. In other eukaryotes (in mammals for example), mitochondria may replicate their DNA and divide mainly in response to the energy needs of the cell, rather than in phase with the cell cycle. When the energy needs of a cell are high, mitochondria grow and divide. When energy use is low, mitochondria are destroyed or become inactive. In such examples mitochondria are apparently randomly distributed to the daughter cells during the division of the cytoplasm. Mitochondrial dynamics, the balance between mitochondrial fusion and fission, is an important factor in pathologies associated with several disease conditions.
The hypothesis of mitochondrial binary fission has relied on the visualization by fluorescence microscopy and conventional transmission electron microscopy (TEM). The resolution of fluorescence microscopy (~200 nm) is insufficient to distinguish structural details, such as double mitochondrial membrane in mitochondrial division or even to distinguish individual mitochondria when several are close together. Conventional TEM has also some technical limitations in verifying mitochondrial division. Cryo-electron tomography was recently used to visualize mitochondrial division in frozen hydrated intact cells. It revealed that mitochondria divide by budding.
An individual's mitochondrial genes are inherited only from the mother, with rare exceptions. In humans, when an egg cell is fertilized by a sperm, the mitochondria, and therefore the mitochondrial DNA, usually come from the egg only. The sperm's mitochondria enter the egg, but do not contribute genetic information to the embryo. Instead, paternal mitochondria are marked with ubiquitin to select them for later destruction inside the embryo. The egg cell contains relatively few mitochondria, but these mitochondria divide to populate the cells of the adult organism. This mode is seen in most organisms, including the majority of animals. However, mitochondria in some species can sometimes be inherited paternally. This is the norm among certain coniferous plants, although not in pine trees and yews. For Mytilids, paternal inheritance only occurs within males of the species. It has been suggested that it occurs at a very low level in humans.
Uniparental inheritance leads to little opportunity for genetic recombination between different lineages of mitochondria, although a single mitochondrion can contain 2–10 copies of its DNA. What recombination does take place maintains genetic integrity rather than maintaining diversity. However, there are studies showing evidence of recombination in mitochondrial DNA. It is clear that the enzymes necessary for recombination are present in mammalian cells. Further, evidence suggests that animal mitochondria can undergo recombination. The data are more controversial in humans, although indirect evidence of recombination exists.
Entities undergoing uniparental inheritance and with little to no recombination may be expected to be subject to Muller's ratchet, the accumulation of deleterious mutations until functionality is lost. Animal populations of mitochondria avoid this buildup through a developmental process known as the mtDNA bottleneck. The bottleneck exploits stochastic processes in the cell to increase the cell-to-cell variability in mutant load as an organism develops: a single egg cell with some proportion of mutant mtDNA thus produces an embryo where different cells have different mutant loads. Cell-level selection may then act to remove those cells with more mutant mtDNA, leading to a stabilization or reduction in mutant load between generations. The mechanism underlying the bottleneck is debated, with a recent mathematical and experimental metastudy providing evidence for a combination of random partitioning of mtDNAs at cell divisions and random turnover of mtDNA molecules within the cell.
DNA repair
Mitochondria can repair oxidative DNA damage by mechanisms analogous to those occurring in the cell nucleus. The proteins employed in mtDNA repair are encoded by nuclear genes, and are translocated to the mitochondria. The DNA repair pathways in mammalian mitochondria include base excision repair, double-strand break repair, direct reversal and mismatch repair. Alternatively, DNA damage may be bypassed, rather than repaired, by translesion synthesis.
Of the several DNA repair process in mitochondria, the base excision repair pathway has been most comprehensively studied. Base excision repair is carried out by a sequence of enzyme-catalyzed steps that include recognition and excision of a damaged DNA base, removal of the resulting abasic site, end processing, gap filling and ligation. A common damage in mtDNA that is repaired by base excision repair is 8-oxoguanine produced by oxidation of guanine.
Double-strand breaks can be repaired by homologous recombinational repair in both mammalian mtDNA and plant mtDNA. Double-strand breaks in mtDNA can also be repaired by microhomology-mediated end joining. Although there is evidence for the repair processes of direct reversal and mismatch repair in mtDNA, these processes are not well characterized.
Lack of mitochondrial DNA
Some organisms have lost mitochondrial DNA altogether. In these cases, genes encoded by the mitochondrial DNA have been lost or transferred to the nucleus.Cryptosporidium have mitochondria that lack any DNA, presumably because all their genes have been lost or transferred. In Cryptosporidium, the mitochondria have an altered ATP generation system that renders the parasite resistant to many classical mitochondrial inhibitors such as cyanide, azide, and atovaquone. Mitochondria that lack their own DNA have been found in a marine parasitic dinoflagellate from the genus Amoebophyra. This microorganism, A. cerati, has functional mitochondria that lack a genome. In related species, the mitochondrial genome still has three genes, but in A. cerati only a single mitochondrial gene — the cytochrome c oxidase I gene (cox1) — is found, and it has migrated to the genome of the nucleus.
Dysfunction and disease
Mitochondrial diseases
Damage and subsequent dysfunction in mitochondria is an important factor in a range of human diseases due to their influence in cell metabolism. Mitochondrial disorders often present as neurological disorders, including autism. They can also manifest as myopathy, diabetes, multiple endocrinopathy, and a variety of other systemic disorders. Diseases caused by mutation in the mtDNA include Kearns–Sayre syndrome, MELAS syndrome and Leber's hereditary optic neuropathy. In the vast majority of cases, these diseases are transmitted by a female to her children, as the zygote derives its mitochondria and hence its mtDNA from the ovum. Diseases such as Kearns-Sayre syndrome, Pearson syndrome, and progressive external ophthalmoplegia are thought to be due to large-scale mtDNA rearrangements, whereas other diseases such as MELAS syndrome, Leber's hereditary optic neuropathy, MERRF syndrome, and others are due to point mutations in mtDNA.
It has also been reported that drug tolerant cancer cells have an increased number and size of mitochondria which suggested an increase in mitochondrial biogenesis. A 2022 study in Nature Nanotechnology has reported that cancer cells can hijack the mitochondria from immune cells via physical tunneling nanotubes.
In other diseases, defects in nuclear genes lead to dysfunction of mitochondrial proteins. This is the case in Friedreich's ataxia, hereditary spastic paraplegia, and Wilson's disease. These diseases are inherited in a dominance relationship, as applies to most other genetic diseases. A variety of disorders can be caused by nuclear mutations of oxidative phosphorylation enzymes, such as coenzyme Q10 deficiency and Barth syndrome. Environmental influences may interact with hereditary predispositions and cause mitochondrial disease. For example, there may be a link between pesticide exposure and the later onset of Parkinson's disease. Other pathologies with etiology involving mitochondrial dysfunction include schizophrenia, bipolar disorder, dementia, Alzheimer's disease, Parkinson's disease, epilepsy, stroke, cardiovascular disease, chronic fatigue syndrome, retinitis pigmentosa, and diabetes mellitus.
Mitochondria-mediated oxidative stress plays a role in cardiomyopathy in type 2 diabetics. Increased fatty acid delivery to the heart increases fatty acid uptake by cardiomyocytes, resulting in increased fatty acid oxidation in these cells. This process increases the reducing equivalents available to the electron transport chain of the mitochondria, ultimately increasing reactive oxygen species (ROS) production. ROS increases uncoupling proteins (UCPs) and potentiate proton leakage through the adenine nucleotide translocator (ANT), the combination of which uncouples the mitochondria. Uncoupling then increases oxygen consumption by the mitochondria, compounding the increase in fatty acid oxidation. This creates a vicious cycle of uncoupling; furthermore, even though oxygen consumption increases, ATP synthesis does not increase proportionally because the mitochondria are uncoupled. Less ATP availability ultimately results in an energy deficit presenting as reduced cardiac efficiency and contractile dysfunction. To compound the problem, impaired sarcoplasmic reticulum calcium release and reduced mitochondrial reuptake limits peak cytosolic levels of the important signaling ion during muscle contraction. Decreased intra-mitochondrial calcium concentration increases dehydrogenase activation and ATP synthesis. So in addition to lower ATP synthesis due to fatty acid oxidation, ATP synthesis is impaired by poor calcium signaling as well, causing cardiac problems for diabetics.
Relationships to aging
There may be some leakage of the electrons transferred in the respiratory chain to form reactive oxygen species. This was thought to result in significant oxidative stress in the mitochondria with high mutation rates of mitochondrial DNA. Hypothesized links between aging and oxidative stress are not new and were proposed in 1956, which was later refined into the mitochondrial free radical theory of aging. A vicious cycle was thought to occur, as oxidative stress leads to mitochondrial DNA mutations, which can lead to enzymatic abnormalities and further oxidative stress.
A number of changes can occur to mitochondria during the aging process. Tissues from elderly humans show a decrease in enzymatic activity of the proteins of the respiratory chain. However, mutated mtDNA can only be found in about 0.2% of very old cells. Large deletions in the mitochondrial genome have been hypothesized to lead to high levels of oxidative stress and neuronal death in Parkinson's disease. Mitochondrial dysfunction has also been shown to occur in amyotrophic lateral sclerosis.
Since mitochondria cover a pivotal role in the ovarian function, by providing ATP necessary for the development from germinal vesicle to mature oocyte, a decreased mitochondria function can lead to inflammation, resulting in premature ovarian failure and accelerated ovarian aging. The resulting dysfunction is then reflected in quantitative (such as mtDNA copy number and mtDNA deletions), qualitative (such as mutations and strand breaks) and oxidative damage (such as dysfunctional mitochondria due to ROS), which are not only relevant in ovarian aging, but perturb oocyte-cumulus crosstalk in the ovary, are linked to genetic disorders (such as Fragile X) and can interfere with embryo selection.
History
The first observations of intracellular structures that probably represented mitochondria were published in 1857, by the physiologist Albert von Kolliker.Richard Altmann, in 1890, established them as cell organelles and called them "bioblasts". In 1898, Carl Benda coined the term "mitochondria" from the Greek μίτος, mitos, "thread", and χονδρίον, chondrion, "granule".Leonor Michaelis discovered that Janus green can be used as a supravital stain for mitochondria in 1900. In 1904, Friedrich Meves, made the first recorded observation of mitochondria in plants in cells of the white waterlily, Nymphaea alba and in 1908, along with Claudius Regaud, suggested that they contain proteins and lipids. Benjamin F. Kingsbury, in 1912, first related them with cell respiration, but almost exclusively based on morphological observations. In 1913, particles from extracts of guinea-pig liver were linked to respiration by Otto Heinrich Warburg, which he called "grana". Warburg and Heinrich Otto Wieland, who had also postulated a similar particle mechanism, disagreed on the chemical nature of the respiration. It was not until 1925, when David Keilin discovered cytochromes, that the respiratory chain was described.
In 1939, experiments using minced muscle cells demonstrated that cellular respiration using one oxygen molecule can form four adenosine triphosphate (ATP) molecules, and in 1941, the concept of the phosphate bonds of ATP being a form of energy in cellular metabolism was developed by Fritz Albert Lipmann. In the following years, the mechanism behind cellular respiration was further elaborated, although its link to the mitochondria was not known. The introduction of tissue fractionation by Albert Claude allowed mitochondria to be isolated from other cell fractions and biochemical analysis to be conducted on them alone. In 1946, he concluded that cytochrome oxidase and other enzymes responsible for the respiratory chain were isolated to the mitochondria. Eugene Kennedy and Albert Lehninger discovered in 1948 that mitochondria are the site of oxidative phosphorylation in eukaryotes. Over time, the fractionation method was further developed, improving the quality of the mitochondria isolated, and other elements of cell respiration were determined to occur in the mitochondria.
The first high-resolution electron micrographs appeared in 1952, replacing the Janus Green stains as the preferred way to visualize mitochondria. This led to a more detailed analysis of the structure of the mitochondria, including confirmation that they were surrounded by a membrane. It also showed a second membrane inside the mitochondria that folded up in ridges dividing up the inner chamber and that the size and shape of the mitochondria varied from cell to cell.
The popular term "powerhouse of the cell" was coined by Philip Siekevitz in 1957.
In 1967, it was discovered that mitochondria contained ribosomes. In 1968, methods were developed for mapping the mitochondrial genes, with the genetic and physical map of yeast mitochondrial DNA completed in 1976.
See also
General
-
 This article incorporates public domain material from Science Primer. NCBI. Archived from the original on 2009-12-08.
This article incorporates public domain material from Science Primer. NCBI. Archived from the original on 2009-12-08.
External links
- Lane N (2016). The Vital Question: Energy, Evolution, and the Origins of Complex Life. WW Norton & Company. ISBN 978-0393352979.
- Powering the Cell Mitochondria – XVIVO Scientific Animation
- Mitodb.com – The mitochondrial disease database.
- Mitochondria Atlas at University of Mainz
- Mitochondria Research Portal at mitochondrial.net
- Mitochondria: Architecture dictates function at cytochemistry.net
- Mitochondria links at University of Alabama
- MIP Mitochondrial Physiology Society
- 3D structures of proteins from inner mitochondrial membrane at University of Michigan
- 3D structures of proteins associated with outer mitochondrial membrane at University of Michigan
- Mitochondrial Protein Partnership at University of Wisconsin
- MitoMiner – A mitochondrial proteomics database at MRC Mitochondrial Biology Unit
- Mitochondrion – Cell Centered Database
- Mitochondrion Reconstructed by Electron Tomography at San Diego State University
- Video Clip of Rat-liver Mitochondrion from Cryo-electron Tomography