
Peripherally acting μ-opioid receptor antagonists (PAMORAs) are a class of chemical compounds that are used to reverse adverse effects caused by opioids interacting with receptors outside the central nervous system (CNS), mainly those located in the gastrointestinal tract. PAMORAs are designed to specifically inhibit certain opioid receptors in the gastrointestinal tract and with limited ability to cross the blood–brain barrier. Therefore, PAMORAs do not affect the analgesic effects of opioids within the central nervous system.
Discovery and development
Opioid drugs are known to cause opioid-induced constipation (OIC) by inhibiting gastric emptying and decreasing peristaltic waves leading to delayed absorption of medications and more water absorption from the feces. That can result in hard and dry stool and constipation for some patients.
OIC is one of the most common adverse effects caused by opioids, so the discovery of PAMORAs can prevent the effects that often compromise pain management.
Methylnaltrexone bromide was the first medication in the drug class approved by the FDA. It was discovered in 1979 by Leon Goldberg, a pharmacologist at the University of Chicago. Having witnessed the suffering of a dying friend with OIC, Goldberg tested various derivatives of naltrexone, a drug known to block the effects of opioids. His objective was to find a drug that could not pass the blood–brain-barrier, without affecting the analgesic effects of the opioids. After Goldberg died, his colleagues at the university continued to develop the compound. It was approved by the FDA in April 2008, originally for OIC in adult patients with advanced illness and later in adult patients with chronic noncancer pain.
In the late 1970s, Dennis M. Zimmerman and his co-workers from Lilly Research Laboratories, Indiana, did research on structural concepts for narcotic antagonists defined in a 4-phenylpiperidine series. They reported N-methyl-trans-3,4-dimethyl-4-phenylpiperidine to be pure opioid receptor antagonist with a new pharmacophore. To increase the potency they attached a phenolic group to the aromatic ring, N-methyl-trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine. That structure was used to design and develop other opioid receptors antagonists such as alvimopan. Alvimopan was approved later in 2008 for in-hospital use to increase the gastrointestinal function following a partial large or small bowel resection with primary anastomosis. Naloxegol was approved in September 2014 and naldemedine in March 2017, both for the treatment of OIC in adult patients with chronic cancer.
Mechanism of action
PAMORAs act by inhibiting the binding of opioids agonist to the μ-opioid receptor (MOR). The objective of PAMORAs treatment is to restore the enteric nervous system function (ENS). The MOR is found in several places in the body and PAMORAs is a competitive antagonist for binding to the receptor. The MORs in the gastrointestinal tract are the main receptors that PAMORAs are intended to block and prevent the binding of opioid agonists. PAMORAs are used in the treatment of opioid-induced bowel dysfunction (OIBD), a potential adverse effect caused by chronic opioid use. PAMORAs act on the three pathophysiological mechanisms of this adverse effect. They act on gut motility, gut secretion and sphincter function.
PAMORAs effect on gut motility is that it can increase the resting tone in the circular muscle layer. The antagonist enhances the effect on tonic inhibition of the muscle tone. This will normalize the tone in the circular muscle layer and therefore prevent opioid-induced rhythmic contractions. When these two factors are combined, it results in decreased transit time. Impliedly these effects will decrease the passive absorption of fluids which helps with decreasing OIBD symptoms such as constipation, gut spasm and abdominal cramp.
PAMORAs effect on gut secretion will help reverse the decreased cAMP formation that opioid agonists induce. Also, the antagonist will establish a normal secretion of chloride. Opioids agonists can also reduce the secretion of peptides by increasing the sympathetic nervous system through the μ-receptors in the ENS, which can lead to drier and harder stool. PAMORAs work against it so the stool becomes softer and less dry.
PAMORAs effect on the function of the sphincter is in theory to regulate the movement coordination. The antagonist can prevent sphincter of Oddi dysfunction that is caused by opioids. Antagonists can also reduce opioid-induced anal sphincter dysfunction. The dysfunction is tied to straining, hemorrhoids and incomplete emptying.
Structure–activity relationship
Even though μ-opioid receptor (MOR) targeting drugs have been used for a long time, not much is known about the structure-activity relationship and the ligand-receptor interactions on the basis of well-defined biological effects on receptor activation or inhibition. Also, the distinction in the receptor-ligand interaction patterns of agonists and antagonists is not known for sure. One theory states that the morphinans biological activity could be determined by the size of the N-substituents. For example, antagonists usually have larger substituents, such as allyl- or cyclopropyl methyl at the morphinan nitrogen, while agonists generally contain a methyl group. On the other hand, agonist activity is also shown in ligands with larger groups at the morphinan nitrogen, and therefore this hypothesis is challenged.
Structure

Methylnaltrexone bromide, naloxegol, and naldemedine all have similar structures, which is not far away from the chemical structure of morphine and other MOR-agonists. All contain a rigid pentacyclic structure that involves benzene ring (A), tetrahydrofuran ring (B), two cyclohexane rings (C and D) and a piperidine ring (E). The most important functional groups for the biological action of opioids are the hydroxyl group on the phenol, N-methyl group, ether bridge between C4 and C5, the double bond between carbon number C7 and C8 and the hydroxyl groups at C3 and C6. The phenolic ring and its 3-hydroxyl group is vital for the analgesic effects as the removal of the OH group decrease the analgesic activity 10-fold. There is another principle for the hydroxyl group on C6 as the removal enhances its activity. The increased activity is mainly because of the increased lipophilicity and the increased ability to cross the blood–brain barrier. Naldemedine has the hydroxyl group while methylnaltrexone bromide has a ketone group and naloxegol has an ester. The double bond between C7 and C8 is not required for the analgesic effect and reduction of the double bond will increase the activity. None of the antagonists has a double bond in their structure. The N-substituent on the skeleton is thought to determine the pharmacological behavior and its interaction with MOR. It is also thought to play a key role in distinguishing antagonists from agonists. Allyl group, a methylcyclopropyl group or a methylcyclobutyl as N-substituent groups are thought to lead antagonist activity.
Binding site
Agonists and antagonists form certain chemical bonds with amino acids that construct the MOR. The majority of antagonists, as well as agonists, are predicted to form charged interaction with Asp147 and a hydrogen bond with Tyr148. However, majority of antagonists also form additional polar interactions with other amino acid residues such as Lys233, Gln124, Gln229, Asn150, Trp318 and Tyr128. Only a small minority of agonists form the same additional polar interactions. Both agonists and antagonists are known to form hydrogen bonds with His297.
It can be concluded that interactions with the amino acid residues, Asp147 and Tyr148 are essential for the ligand to bind to the receptor and the molecules that form additional polar interactions with other residues are more often antagonists than agonists.
The N-substituent group can form hydrophobic bonds with Tyr326 and Trp293 and the aromatic and cyclohexane rings can form similar bonds to Met151. The backside of the ligand can also form a hydrophobic bond, but with Val300 and Ile296.
Methylnaltrexone bromide
Methylnaltrexone bromide is the bromide salt form of methylnaltrexone, a quaternary methyl derivative of noroxymorphone. The methyl group and the quaternary salt formation increase the polarity and reduce the lipid solubility thereby restricts the blood–brain-barrier penetration. Methylnaltrexone has eight times higher affinity for MOR than for κ-opioid receptor (KOR) and δ-opioid receptor (DOR). Naltrexone forms interaction with Asp147 and Tyr148 along with a hydrogen bond with Lys233.

Alvimopan

Peripherally selective trans-3,4-dimethyl-4-(3-hydroxylphenyl)piperidine opioid antagonists were developed for the treatment of gastrointestinal motility disorder by Zimmerman and his coworkers. From that, they derived the 4-(3-hydroxyphenyl)-3,4-dimethylpiperidine scaffold with functional groups spanning various sizes, charge, and polarity to reach peripheral opioid receptor antagonism while decreasing CNS drug exposure. The in vitro μ-Ki, in vivo AD50, and ED50 and peripheral index (ratio) was examined for several selective analogs, and from that, they found out that the trans-3,4-dimethyl-4-(3-hydroxyphenyl) piperidine, Alvimopan, gave the best results. The large zwitterionic structure and the high polarity prevents Alvimopan from crossing the blood–brain barrier, potency at binding peripheral MORs is thereby 200 times that of central MORs.
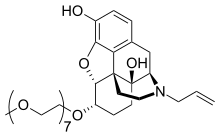
Naloxegol
Naloxegol is a polyethylene glycol-modified derivative of α-naloxol. Naloxegol has a similar form as naloxone as a heteropentacyclic compound both of which have an allyl group attached to the amine of the piperidine ring. However, naloxegol has a monomethoxy-terminated n=7 oligomer of PEG connected to the 6-alpha-hydroxyl group of ɑ-naloxol via an ether linkage. The PEG moiety increases the molecular weight and therefore restricts the uptake of naloxegol into the CNS. Furthermore, pegylated naloxegol becomes a substrate for the P-glycoprotein efflux transporter that transports the compound out of the CNS.
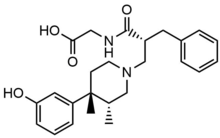
Naldemedine
Naldemedine has a similar chemical structure as naltrexone but with an additional side chain that increases the molecular weight and polar surface area of the substance. Like naloxegol, naldemedine is a substrate of the P-glycoprotein efflux transporter. These properties result in less penetration into the CNS and decrease possible inference with the effects of opioid agonists. Naldemedine is a dual antagonist for MOR and DOR. Activation of the DOR has been known to cause nausea and/or vomiting, so a dual antagonist can decrease both OIC and nausea/vomiting.
Pharmacokinetics
The molecular weight, bioavailability, protein binding, elimination half-life, the time to achieve maximum plasma concentration and binding affinity are present in the table below.
| Chemical name | Chemical structure | Molecular weight (g/mol) | Bioavailability (%) | Plasma protein binding (%) | t1/2 (h) | tmax | Ki μ (nM) | Ki κ (nM) | Ki δ (nM) |
|---|---|---|---|---|---|---|---|---|---|
| Methylnaltrexone bromide |
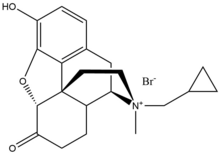 |
436,3 | Low | 11-15 | 8 | 30 min | 5.50 | 32.1 | 3453.8 |
| Alvimopan |
|
424,53 | 6 | 80-90 | 10-17 | 2 h | 0.77 | 40 | 4.4 |
| Naloxegol |
|
651,798 | NA | 4,2 | 6-11 | 2 h | 7.42 | 8.65 | 203.0 |
| Naldemedine |
|
570,6 | 29 | 93-94 | 11 | 45 min | 0.34 | 0.94 | 0.43 |

- t1/2: Biological half-life
- tmax: Time to achieve maximum plasma concentration
- pKi: the measurement of ligand binding affinity
Methylnaltrexone bromide has poor oral bioavailability, and for that reason, every other day it is administered subcutaneously. About half of the dose is excreted in the urine and somewhat less in feces with 85% eliminated unchanged.
Alvimopan has considerable low bioavailability (6%) due to its high binding affinity and low dissociation rate. Essentially, alvimopan is mediated by biliary secretion with an average plasma clearance of 400 ml/min. Metabolism of alvimopan is via intestinal flora resulting in hydrolysis of alvimopan to the active amide metabolite (ADL 08-0011). However, the metabolite is considered clinically irrelevant due to its low binding affinity.
When naloxegol is given with a fatty meal, absorption increases. Clearance is mostly via hepatic metabolism (P450-CYP3A) with unknown actions of the metabolites. Naloxegol has small fragments eliminated by renal excretion.
Naldemedine metabolites mainly via CYP3A to nor-naldemedine, it also metabolites via UDP-glucuronosyltransferase 1A3 to naldemedine 3-G, but in a lesser extent. Those metabolites are both opioid receptor antagonists but are less potent than the parent compound.
PAMORAs in development

Axelopran is an oral PAMORA which is under development by Theravane Biopharma. It has completed phase II in clinical trials in more than 400 patients with OIC. Axelopran has a different chemical structure from other PAMORAs but with a similar mechanism of action. It acts as an antagonist for MOR, KOR and DOR, but with higher affinity for MOR and KOR than for DOR. Like other PAMORAs, the main goal is the treatment of OIC. Axelopran is also being investigated in fixed-dose combination (FDC) with oxycodone. It is done by using spray coating technology to create an FDC of axelopran and controlled-release oxycodone.
There is a demand for optimization of the receptor selectivity and affinity accompanied by an exploration of candidate compounds regarding their route of administration. These are the main objectives and future strategies for drug discovery and the development of PAMORAs. Predominantly, the MORs exhibit functionally selective agonism. Therefore, future possible candidate compounds that target OIC are PAMORAs with optimized selectivity and affinity.