
The pharmacology of progesterone, a progestogen medication and naturally occurring steroid hormone, concerns its pharmacodynamics, pharmacokinetics, and various routes of administration.
Progesterone is a naturally occurring and bioidentical progestogen, or an agonist of the progesterone receptor, the biological target of progestogens like endogenous progesterone. Progesterone also has antimineralocorticoid and inhibitory neurosteroid activity, whereas it appears to have little or no glucocorticoid or antiandrogenic activity and has no androgenic activity. Because of its progestogenic activity, progesterone has functional antiestrogenic effects in certain tissues such as the uterus, cervix, and vagina. In addition, progesterone has antigonadotropic effects due to its progestogenic activity and can inhibit fertility and suppress sex hormone production. Progesterone differs from progestins (synthetic progestogens) like medroxyprogesterone acetate and norethisterone, with implications for pharmacodynamics and pharmacokinetics as well as efficacy, tolerability, and safety.
Progesterone can be taken by mouth, in through the vagina, and by injection into muscle or fat, among other routes. A progesterone vaginal ring and progesterone intrauterine device are also available as pharmaceutical products.
Mechanism of action
Progesterone is a progestogen, or an agonist of the nuclear progesterone receptors (PRs), the PR-A, PR-B, and PR-C. In one study, progesterone showed EC50 values of 7.7 nM for the human PR-A and 8.0 nM for the human PR-B. In addition to the PRs, progesterone is an agonist of the membrane progesterone receptors (mPRs), including the mPRα, mPRβ, mPRγ, mPRδ, and mPRϵ. It is also a potent antimineralocorticoid (antagonist of the mineralocorticoid receptor (MR)), as well as a very weak glucocorticoid (agonist of the glucocorticoid receptor). Progesterone does not interact significantly with the androgen receptor (AR) or with the estrogen receptor (ER). In addition to its activity as a steroid hormone, progesterone is a neurosteroid. Specifically, it is an antagonist of the sigma σ1 receptor, a negative allosteric modulator of nicotinic acetylcholine receptors, and, via its active metabolites allopregnanolone and pregnanolone, a potent positive allosteric modulator of the GABAA receptor, the major signaling receptor of the inhibitory neurotransmitter γ-aminobutyric acid (GABA).
| Compound | PR | AR | ER | GR | MR | SHBG | CBG |
|---|---|---|---|---|---|---|---|
| Progesterone | 50 | 0 | 0 | 10 | 100 | 0 | 36 |
| Notes: Values are percentages (%). Reference ligands (100%) were promegestone for the PR, metribolone for the AR, E2 for the ER, DEXA for the GR, aldosterone for the MR, DHT for SHBG, and cortisol for CBG. Sources: | |||||||
| Compound | Form | Dose for specific uses (mg) | DOA | |||
|---|---|---|---|---|---|---|
| TFD | POICD | CICD | ||||
| Algestone acetophenide | Oil soln. | - | – | 75–150 | 14–32 d | |
| Gestonorone caproate | Oil soln. | 25–50 | – | – | 8–13 d | |
| Hydroxyprogest. acetate | Aq. susp. | 350 | – | – | 9–16 d | |
| Hydroxyprogest. caproate | Oil soln. | 250–500 | – | 250–500 | 5–21 d | |
| Medroxyprog. acetate | Aq. susp. | 50–100 | 150 | 25 | 14–50+ d | |
| Megestrol acetate | Aq. susp. | - | – | 25 | >14 d | |
| Norethisterone enanthate | Oil soln. | 100–200 | 200 | 50 | 11–52 d | |
| Progesterone | Oil soln. | 200 | – | – | 2–6 d | |
| Aq. soln. | ? | – | – | 1–2 d | ||
| Aq. susp. | 50–200 | – | – | 7–14 d | ||
|
Notes and sources:
| ||||||
Antimineralocorticoid activity
Progesterone is a potent antimineralocorticoid. It has 1000% of the affinity of aldosterone, the major endogenous agonist, for the human MR, and 100% of the affinity of aldosterone for the rat MR. Progesterone produces antimineralocorticoid effects such as natriuresis (excretion of sodium in the urine) at normal physiological concentrations. A 200 mg dose of oral progesterone is considered to be approximately equivalent in antimineralocorticoid effect to a 25 to 50 mg dose of the potent antimineralocorticoid spironolactone, which itself is a derivative of progesterone. Doses of progesterone of 50 to 200 mg by intramuscular injection, which are similar to progesterone exposure in the third trimester of pregnancy, have also been reported to produce antimineralocorticoid-like effects. The antimineralocorticoid effects of progesterone underlie its ability to lower blood pressure and reduce water and salt retention and its potential application in the treatment of hypertension. An active metabolite of progesterone, 11-deoxycorticosterone (21-hydroxyprogesterone), is a precursor of aldosterone and has strong mineralocorticoid activity (i.e., is a strong agonist of the MR). However, it is formed in relatively small amounts, and any such effects produced by it are usually outweighed by the antimineralocorticoid activity of progesterone. Progesterone may be a relatively weak antimineralocorticoid in vivo.
Glucocorticoid activity
Progesterone is a partial agonist of the glucocorticoid receptor (GR). It has about 35% of the affinity of dexamethasone, a corticosteroid, for the human GR, and about 3 to 11% of the affinity of dexamethasone for the rat GR. However, progesterone appears to show weak or no glucocorticoid activity and no antiglucocorticoid activity in vitro and in animals. Nonetheless, progesterone has been found to upregulate the thrombin receptor in vascular smooth muscle cells in vitro, a glucocorticoid effect, and this could have clinical relevance in relation to risk of blood clots.
| Steroid | Class | TR (↑)a | GR (%)b |
|---|---|---|---|
| Dexamethasone | Corticosteroid | ++ | 100 |
| Ethinylestradiol | Estrogen | – | 0 |
| Etonogestrel | Progestin | + | 14 |
| Gestodene | Progestin | + | 27 |
| Levonorgestrel | Progestin | – | 1 |
| Medroxyprogesterone acetate | Progestin | + | 29 |
| Norethisterone | Progestin | – | 0 |
| Norgestimate | Progestin | – | 1 |
| Progesterone | Progestogen | + | 10 |
| Footnotes: a = Thrombin receptor (TR) upregulation (↑) in vascular smooth muscle cells (VSMCs). b = RBA (%) for the glucocorticoid receptor (GR). Strength: – = No effect. + = Pronounced effect. ++ = Strong effect. Sources: | |||
Androgenic and antiandrogenic activity
The binding and activity of progesterone at the AR, the biological target of androgens like testosterone and dihydrotestosterone (DHT) in the body, is controversial. Some studies have found progesterone to bind to the AR, with agonistic and antagonistic activity exerted, whereas other studies have found very low or no affinity for the AR at all. In animal studies, no androgenic effects have been observed, but weak antiandrogenic effects have been reported. The weak antiandrogenic activity has been attributed not to antagonism of the AR by progesterone, but rather to its weak 5α-reductase inhibition and consequent inhibition of the conversion of testosterone into the more potent DHT. There is no clinical evidence of AR-mediated androgenic or antiandrogenic activity with progesterone. Progesterone has not been associated with any classical androgenic effects in clinical studies in women, including no changes in the blood lipid profile or sex hormone-binding globulin levels, acne, oily skin, hirsutism, or voice deepening, nor with virilization of female fetuses. As such, the scientific consensus is that progesterone is clinically neither androgenic nor antiandrogenic. This is in contrast to many progestins, such as 19-nortestosterone derivatives (e.g., norethisterone, levonorgestrel, dienogest) and 17α-hydroxyprogesterone derivatives (e.g., cyproterone acetate, medroxyprogesterone acetate), which do bind to the AR and have been associated with significant androgenic or antiandrogenic effects depending on the progestin in question. Due to its lack of androgenic and antiandrogenic activity, and hence lack of masculinizing and feminizing effects, progesterone is one of the few progestogens that is suitable for use during pregnancy in women at risk for preterm birth or recurrent miscarriage.
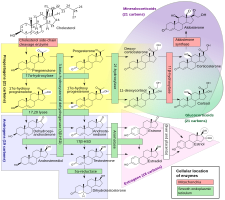
Although progesterone does not have significant AR-mediated androgenic or antiandrogenic activity, it is a precursor and intermediate, albeit distant, in the biosynthesis of androgens from cholesterol. For this reason, there has been some speculation that exogenous progesterone could be transformed into androgens by certain tissues that express the requisite enzymes. Progesterone is converted by 17α-hydroxylase into 17α-hydroxyprogesterone; 17α-hydroxyprogesterone is converted by 17,20-lyase into androstenedione; and androstenedione is converted by 17β-hydroxysteroid dehydrogenases into testosterone.CYP17A1, the cytochrome P450 gene that encodes 17α-hydroxylase and 17,20-lyase, is expressed mainly in the gonads (ovaries and testes) and the adrenal glands. However, while it is theoretically possible that progesterone could be transformed in the body into androgens, no androgenic effects have been observed in animal studies. In addition, clinical studies, in which women were treated with 100 to 300 mg/day oral progesterone, have found no or only a small increase in levels of 17α-hydroxyprogesterone, and no change in androgen levels, including of dehydroepiandrosterone, androstenedione, and testosterone. In these studies, levels of estradiol and cortisol, which progesterone is also a precursor of, did not change either, although levels of 11-deoxycorticosterone increased significantly. Levels of androgens, like testosterone and dihydrotestosterone (DHT), also do not increase going from the follicular phase to the luteal phase of the menstrual cycle in premenopausal women (progesterone levels being high in the luteal phase).
5α-Reductase inhibition
Progesterone is a substrate for 5α-reductase, and has been found to act as a competitive inhibitor of this enzyme in vitro in a variety of studies. In one study, it showed IC50 values of 1,375 nM and 88 nM (in the presence of 50 nM androstenedione as the substrate) for 5α-reductase types 1 and 2, respectively. 5α-Reductase is highly expressed in skin, hair follicles, and prostate gland, and is responsible for the transformation of testosterone into the several-fold more potent androgen DHT in such tissues. As such, it has been suggested that progesterone might possess some antiandrogenic effect via acting as a 5α-reductase inhibitor. However, inhibition of 5α-reductase by progesterone is described as a weak effect that has only been demonstrated in vitro and at supraphysiological concentrations. In accordance, physiological levels of circulating progesterone have not been found to importantly influence circulating DHT concentrations.
Congenital 5α-reductase 2 deficiency is a rare intersex condition which is associated with ambiguous genitalia in male fetuses due to a deficiency in DHT production during genital differentiation. Experimental prenatal exposure to established 5α-reductase inhibitors like finasteride has been found to produce similar feminized genital defects in male animals including rodents and monkeys. In contrast, exogenous administration of progesterone to pregnant rodents and monkeys has resulted in minimal abnormality in either male or female pups. In addition, endogenous progesterone levels naturally increase to extremely high concentrations during pregnancy, yet genital defects do not occur. In accordance, while total concentrations of progesterone in pregnant women at term are around 150 ng/mL (~500 nM), free or unbound and hence bioactive concentrations of progesterone are only about 3 ng/mL (~10 nM) due to the high plasma protein binding of progesterone, and these concentrations are still well below the aforementioned IC50 values for inhibition of 5α-reductase types 1 and 2. As with endogenous progesterone during pregnancy, exogenous supplemental progesterone during pregnancy has been found not to increase the risk of hypospadias in male infants.
Although systemic progesterone does not appear to be an effective 5α-reductase inhibitor, topical progesterone may produce potent inhibition of 5α-reductase in the skin due to the very high local concentrations that occur. A study found that topical progesterone applied to the pubic area in men inhibited 5α-reductase in the skin in this region by 75%. In addition to inhibition of 5α-reductase, progesterone is metabolized by 5α-reductase into 5α-dihydroprogesterone (5α-DHP), a compound reported to have some antagonistic activity at the AR. However, this compound appears to have no systemic antiandrogenic activity. In spite of its apparent 5α-reductase inhibition, the effectiveness of topical progesterone in the treatment of pattern hair loss has been poor.
Other activity
Certain progestins have been found to stimulate the proliferation of MCF-7 breast cancer cells in vitro, an action that is independent of the classical PRs and is instead mediated via the progesterone receptor membrane component-1 (PGRMC1). Progesterone, nomegestrol acetate, and chlormadinone acetate act neutrally and do not stimulate proliferation, whereas norethisterone, desogestrel, levonorgestrel, and drospirenone strongly stimulate proliferation and medroxyprogesterone acetate, dienogest, and dydrogesterone weakly stimulate proliferation. As such, progesterone differs from some but not all progestins in the activity mediating this PGRMC1-dependent effect. It is unclear if these findings may explain the different risks of breast cancer observed with progesterone, dydrogesterone, and other progestins such as medroxyprogesterone acetate and norethisterone in clinical studies.
Effects in the body and brain
The PRs are expressed widely throughout the body, including in the uterus, cervix, vagina, fallopian tubes, breasts, fat, skin, pituitary gland, hypothalamus, and elsewhere throughout the brain. Through activation of the PRs (as well as the mPRs), progesterone has many effects, including the following:
- Induces endometrial secretory transformation in preparation for pregnancy (>5 ng/mL)
- Prevents estrogen-induced endometrial hyperplasia and increased endometrial cancer risk
- Maintains pregnancy via effects in endometrium (with withdrawal resulting in miscarriage)
- Reduces amount and fibrosity of cervical mucus and causes cervix to become firmer and more tightly closed
- Controls motility and composition of fluid in the fallopian tubes
- Reduced cornification and maturation of the vaginal lining
- Causes water retention in the breasts resulting in temporary enlargement during the menstrual cycle
- Mediates lobuloalveolar development of the breasts necessary for lactation
- Suppresses lactation initiation and triggers lactation upon withdrawal (as with parturition)
- Maintains skin health, integrity, appearance, and hydration and slows the rate of aging of the skin
- Modulates brain function, with effects on mood, emotionality, and sexuality, as well as cognition and memory
- Exerts negative feedback on the hypothalamic–pituitary–gonadal axis (HPG axis) by suppressing the secretion of the gonadotropins FSH and LH from the pituitary gland (including the mid-cycle gonadotropin surge), thereby inhibiting gonadal sex hormone production as well as ovulation and fertility (>2 ng/mL)
- Increases basal body temperature (by 0.3–0.6 °C (0.5–1.0 °F) relative to preovulation) via the hypothalamus (>4 ng/mL)
- Reduces hot flashes via the hypothalamus
- Stimulates respiration via the hypothalamus and/or respiratory center
- Influences the risk and/or progression of hormone-sensitive cancers including breast cancer and endometrial cancer
Many of the effects of progesterone require estrogen, as estrogens prime tissues for progesterone by inducing expression of the PRs. The PRs are induced in the breasts by estrogens, and for this reason, it is assumed that progestogens cannot mediate breast changes in the absence of estrogens.
Progesterone also lowers blood pressure and reduces water and salt retention among other effects via its antimineralocorticoid activity.
Progesterone can produce sedative, hypnotic, anxiolytic, euphoric, cognitive-, memory-, and motor-impairing, anticonvulsant, and even anesthetic effects via formation of sufficiently high concentrations of its neurosteroid metabolites and consequent GABAA receptor potentiation in the brain.
Uterine effects
Under normal physiological circumstances, progesterone secreted by the corpus luteum during the luteal phase of the menstrual cycle produces endometrial transformation of the estrogen-primed uterus in preparation for implantation and pregnancy. Normal progesterone production during the luteal phase is 25 mg/day on average with a range of 15 to 50 mg/day. Progesterone levels during the luteal phase range from 7 ng/mL to 22 ng/mL using liquid chromatography–tandem mass spectrometry (LC–MS/MS) per one source. Sustained progesterone levels of more than 5 ng/mL, perhaps approximately 10 ng/mL, are required for full endometrial transformation. Progesterone levels of more than 10 ng/mL are rarely associated with luteal-phase defect on the basis of endometrial biopsy.
Luteal-phase levels of progesterone are said to be produced by 25 mg/day progesterone in oil solution by intramuscular injection or by 100 mg/day progesterone by vaginal or rectal administration. Progesterone by intramuscular injection in oil solution has been found to produce endometrial transformation at a dose of 10 or 20 mg/day for 14 days (total dose per cycle of 200 mg), whereas a single intramuscular injection of 200 mg progesterone in microcrystalline aqueous suspension provides endometrial transformation after 10 to 14 days. A study found full and equivalent endometrial transformation with subcutaneous injection of 25 mg/day versus 50 mg/day progesterone in aqueous solution. Due to a uterine first-pass effect and markedly higher uterine progesterone levels than with other routes, 45 mg/day vaginal progesterone, a dosage that achieves circulating progesterone levels of only 1 to 5 ng/mL, provides complete endometrial transformation. Conversely, intranasal administration of progesterone achieving progesterone levels of 2 to 5 ng/mL was ineffective. Transdermal progesterone achieves very low progesterone levels and is considered to be ineffective for endometrial protection.
The endometrial transformation dosage of oral micronized progesterone in women has been listed as 200 to 300 mg/day or 4,200 mg total per cycle. However, a clinical study found that 300 mg/day oral micronized progesterone was insufficient for full endometrial transformation. Similarly, 600 to 1,000 mg/day oral micronized progesterone has been reported to be ineffective for achieving complete endometrial transformation. Despite inadequate endometrial transformation with oral progesterone, continuous 100 mg/day oral micronized progesterone or cyclic 200 mg/day oral micronized progesterone is effective for protection of the endometrium against estrogen-induced endometrial hyperplasia. On the other hand, and in contrast to progestins, typical clinical doses of oral micronized progesterone have been associated with failure to prevent increased endometrial cancer risk caused by estrogen therapy.
Antiestrogenic effects
Progesterone, like all progestogens, has antiestrogenic effects in certain tissues such as the uterus, cervix, vagina, and breasts, and possibly also the brain. These effects are mediated by activation of the PR in these tissues. Progesterone does not have antiestrogenic effects in the more conventional sense of binding to and antagonizing the ER or binding to and inhibiting enzymes involved in estrogen biosynthesis. Instead, for instance in the endometrium, progesterone causes downregulation of the ER and upregulation of the estrogen-inactivating enzymes 17β-hydroxysteroid dehydrogenase 2 (converts estradiol into estrone) and estrone sulfotransferase (converts estrone into estrone sulfate). The antiestrogenic effects of progesterone and other progestogens form the basis for their only approved indication in menopausal hormone therapy: prevention of long-term unopposed estrogen-induced endometrial hyperplasia and increased endometrial cancer risk in women with intact uteruses.
In the breasts, progesterone and other progestogens downregulate the ER as well as the estrogen-activating enzymes steroid sulfatase (converts estrone sulfate into estrone) and 17β-hydroxysteroid-dehydrogenase 1 (converts estrone into estradiol) and upregulates estrone sulfotransferase. However, other studies suggest that progestogens do not downregulate ER expression in the breasts. When applied directly to the breasts in women, progesterone can block the proliferative effects of estradiol. However, the concentrations were supraphysiological and the same may not be the case with more physiological concentrations. Cellular proliferation in the breasts is greatest in the luteal phase of the menstrual cycle, when progesterone levels are highest.
It has been hypothesized that progestogens may counteract various effects of estrogens in the brain such as stimulatory and excitatory effects on neuronal activity. Progesterone moreover has a special position among progestogens concerning such actions due to its inhibitory neurosteroid metabolites and their central depressant effects. It has been suggested that these actions of progestogens may explain unfavorable effects on mood that have been reported with these medications in some women. However, the mutual interactions of estrogens and progestogens in the brain in general are controversial and require more research.
Progesterone can produce body-wide antiestrogenic effects at very high doses in both women and men via its antigonadotropic effects and consequent suppression of gonadal estrogen production (see below). These antigonadotropic effects are mediated by hyperactivation of the PR.
Effects on the HPG axis
Antigonadotropic effects
Progestogens have antigonadotropic effects at sufficiently high doses via activation of the PR and consequent negative feedback on and hence suppression of the hypothalamic–pituitary–gonadal axis (HPG axis). This results in suppression of gonadotropin secretion and by extension interference with fertility and gonadal sex hormone production. Progesterone prevents ovulation by suppressing the mid-cycle surge in gonadotropin secretion during the menstrual cycle.
The ovulation-inhibiting (i.e., contraceptive) dosage of oral crystalline (non-micronized) progesterone in women is 300 mg/day or greater. However, this figure is based on limited clinical data. In the clinical research in the 1950s that determined this dosage, ovulation inhibition occurred in 50 to 100% of women when assessed via measures including urinary pregnanediol excretion, daily basal body temperatures, endometrial biopsies, and vaginal smears. Another study found that ovulation inhibition with 300 mg/day oral non-micronized progesterone occurred in a "proportion of the cases" when assessed via laparotomy. A third study found that ovulation was inhibited in only 38% of women treated with 1,000 mg/day oral non-micronized progesterone. A fourth publication stated that even 750 to 1,000 mg/day oral non-micronized progesterone had weak effects as evidenced by poor thermogenic effect, weak endometrial effect, and lack of production of withdrawal bleeding in amenorrheic women. Neumann and colleagues listed the ovulation-inhibiting dosage of oral non-micronized progesterone in women as 300 to 500 mg/day or as 400 mg/day but provided no other details.
In a study of a progesterone vaginal ring alone or in combination with estradiol that released 1.5 to 3 mg/day progesterone and achieved mean progesterone levels varying between 0.7 and 1.6 ng/mL (mean 0.9 ng/mL) during anovulatory cycles, ovulation occurred in 18 of 30 (60%) menstrual cycles. A study of a vaginal progesterone ring that released almost 10 mg/day progesterone and maintained mean progesterone levels of 4.4 ng/mL (range 2.4–6.5 ng/mL) found that ovulation was inhibited in some but not all women. In another study, a progesterone vaginal ring that released about 10 mg/day progesterone and produced progesterone levels of around 4 ng/mL (range 3–5.2 ng/mL) resulted in ovulation occurring in 25% of treated breastfeeding women compared to a rate of 56% in a control group of breastfeeding women. A study in rhesus monkeys found that a vaginal ring delivering 0.235 or 1.77 mg/day progesterone inhibited ovulation in all monkeys at the higher dose and in a proportion of monkeys at the lower dose. A dose of progesterone of 5 to 10 mg/day by intramuscular injection has been found to prevent ovulation in women and has been considered effective as a progestogen-only injectable contraceptive.
Short-term therapy with 300 mg/day oral progesterone had no effect on luteinizing hormone pulse frequency in women. Treatment with a high dosage of oral progesterone of 100 mg four times per day (or 400 mg/day total) in men for 10 days did not cause any change in testosterone levels, suggesting that oral progesterone has little or no antigonadotropic effect in males at typical clinical dosages. In addition, a study found that administration of 1,000 mg/day oral progesterone for 3 months had no significant effect on urinary gonadotropin excretion. On the other hand, a single 50 mg intramuscular injection of progesterone, which is associated with high progesterone levels of approximately 50 ng/mL (or early- to mid-pregnancy levels), resulted in substantial (50–60%) suppression of luteinizing hormone, follicle-stimulating hormone, and testosterone levels in men. Similarly, continuous or intermittent intravenous injections of 100 to 400 mg/day progesterone for 10 days significantly decreased urinary gonadotropin excretion. Progestogens in general are able to suppress gonadal testosterone production in men by a maximum of about 70 to 80% or to just above castrate levels when used at sufficiently high doses.
A study using 50 mg/day progesterone by intramuscular injection in five men found that the medication produced azoospermia or severe oligozoospermia in all within 10 weeks, suppressed libido, erectile function, and ejaculate volume to minimal levels, produced slight gynecomastia in two of the men, moderately decreased testicular size, and impaired testicular morphology. Upon discontinuation, sperm counts returned to normal in the men within 14 to 17 weeks. In another study, 100 mg rectal suppositories of progesterone given five times per day for 9 days resulted in progesterone levels of 5.5 to 29 ng/mL and suppressed circulating testosterone and growth hormone levels by about 50% in men, but did not affect libido or erectile potency with this short duration of therapy.
Progonadotropic effects
Progesterone can have progonadotropic effects under certain circumstances.
Neurosteroid effects
Progesterone, through the actions of neurosteroid active metabolites such as allopregnanolone and pregnanolone, is a potent positive allosteric modulator of the GABAA receptor, the major signaling receptor of the inhibitory neurotransmitter γ-aminobutyric acid (GABA). It can produce sedative, hypnotic, anxiolytic, euphoric, cognitive-, memory-, and motor-impairing, anticonvulsant, and even anesthetic effects with formation of sufficiently high concentrations of its neurosteroid metabolites and consequent GABAA receptor potentiation in the brain. These actions and effects are characteristically similar to those of other GABAA receptor positive allosteric modulators like alcohol, barbiturates, and benzodiazepines.
Similarly to other GABAA receptor positive allosteric modulators like alcohol, barbiturates, and benzodiazepines, tolerance has been found to develop with exposure to increased levels of allopregnanolone and related inhibitory neurosteroids. This includes downregulation and desensitization of the GABAA receptor, reduced effects of allopregnanolone and other GABAA receptor activators (e.g., GABA and benzodiazepines), and rebound or withdrawal effects upon falls in allopregnanolone levels. In addition, changes in allopregnanolone levels have been implicated in adverse neuropsychiatric effects associated with the menstrual cycle (e.g., dysphoria, depression, anxiety, irritability) and postpartum period (e.g., postpartum depression), as well as in catamenial epilepsy (seizures). Low and high levels of allopregnanolone seem to have a neutral effect on mood, whereas moderate levels have a negative effect, which may underlie the symptoms of premenstrual syndrome and premenstrual dysphoric disorder that are observed in 30 to 40% of premenopausal women. This U-shaped effect on mood appears to be a common property of GABAA receptor positive allosteric modulators.
See also
Further reading
- Sitruk-Ware R, Bricaire C, De Lignieres B, Yaneva H, Mauvais-Jarvis P (October 1987). "Oral micronized progesterone. Bioavailability pharmacokinetics, pharmacological and therapeutic implications--a review". Contraception. 36 (4): 373–402. doi:10.1016/0010-7824(87)90088-6. PMID 3327648.
- Simon JA (December 1995). "Micronized progesterone: vaginal and oral uses". Clinical Obstetrics and Gynecology. 38 (4): 902–14. doi:10.1097/00003081-199538040-00024. PMID 8616985.
- Ruan X, Mueck AO (November 2014). "Systemic progesterone therapy--oral, vaginal, injections and even transdermal?". Maturitas. 79 (3): 248–55. doi:10.1016/j.maturitas.2014.07.009. PMID 25113944.